
Antimalarial activity of a novel series of artemisinin-derived 1, 2,3-triazole dimers
2019-06-11 02:04:32KabitaGogoiGokulBaishyaBiswajitSaikiaNabinChandraBaruaChandrajitDohutiaAkaleshKumarVermaAnilPrakash
Kabita Gogoi, Gokul Baishya, Biswajit Saikia, Nabin Chandra Barua, Chandrajit Dohutia,4,Akalesh Kumar Verma, Anil Prakash,6✉
1ICMR-Regional Medical Research Centre, N.E., Dibrugarh-786 001, Assam, India
2CSIR-North-East Institute of Science and Technology, Jorhat-785 001, Assam, India
3Digboi College, Chemistry Department, Tinsukia-786 171, Assam, India
4Pratiksha Institute of Pharmaceutical Scinces, Panikheti, Guwahati -781 026, Assam, India
5Cotton University, Zoology Department, Guwahati-781 001, Assam, India
6ICMR-National Institute for Research in Environmental Health, Bhopal-462 001, Madhya Pradesh, India
Keywords:Antimalarial activity Artemisinin derivatives Huisgen reaction Triazole dimers Plasmodium berghei Plasmodium falciparum Molecular docking
ABSTRACT Objective: To obtain suitable artimisinin-based drug candidates with high antimalarial activity.Methods: Three different reaction schemes were used to synthesize a total of 15 artemisininbased compounds. The first synthetic scheme involved the synthesis of diazido aliphatic and aromatic compounds from commercially available dihalides and azido derivatives of artemisinin. The second scheme consisted of the reaction of dibromoaliphatic compounds with sodium azide in dimethylformamide which yielded the desired compounds. Artemisinin-based compounds on treatment with sodium azide and bromotrimethylsilane in dichloromethane produced the most potent compound GB-2. Another potent compound GB-1 was synthesized from artemisinin by treatment with alcohols in the presence of Aberlyst-15 in anhydrous dichloromethane. The third scheme involved the Huisgen 1,3-dipolar cycloaddition between the synthesized aliphatic and aromatic diazides and two alkyne derivatives of artemisinin to obtain the desired artemisinin dimers with average yields.Results: The best in vitro antiplasmodial activity was shown by the compound GB-2 registering IC50 value 0.066 μg/mL against chloroquine-sensitive and 0.865 μg/mL against chloroquineresistant strains of Plasmodium falciparum. It suppressed 59.0% parasitaemia in vivo of rodent malaria parasite Plasmodium berghei in Swiss albino model at 50 μg/kg body weight dosage.Molecular docking interactions of Plasmodium falciparum ATP6 (PfATP6) protein revealed strong bonding of GB-2 with Thr255 residue which is likely to be the reason for excellent antimalarial activity of this compound.Conclusion: Two compounds GB-1 and GB-2 exhibited excellent in vitro antiplasmodial activity and fair in vivo antimalarial activity. Of the two, GB-2 showed better activity which could be attributed to its strong bonding interactions with Thr255 as evidenced from the molecular docking study. Study helped in identifying artemisinin analogues possessing good antimalarial properties and further research in structural alterations of the selected molecules should be carried out which may result in obtaining potent drug candidates against the malarial parasite.
1. Introduction
Malaria has been a life threatening disease for thousands of years and still threatens millions of lives every year, particularly in Sub Saharan African countries[1]. An estimated 216 million cases of malaria occurred worldwide in 2016, of which, majority of the cases were reported from the WHO African Region, followed by the South-east Asia Region and the Eastern Mediterranean Region.The global incidence rate of malaria has decreased by an estimated figure of 18% between the years 2010 and 2016. Despite this overall decrease, substantial increases in case incidence have been reported in the WHO region of the Americas, and marginally in Southeast Asia, Western Pacific and African regions between 2014 and 2016. Most of the estimated malaria cases in sub-Saharan Africa in 2016 have been caused by Plasmodium (P.) falciparum while Plasmodium vivax is the predominant parasite in the WHO Region of the Americas, representing 64% of malaria cases, which is way above 30% cases in the WHO South-east Asia and 40% in the Eastern Mediterranean regions[2]. The emergence of artemisinin and its derivatives as a new class of antimalarial drugs in the early 1970s appeared to be the best development for the treatment of severe malaria caused by P. falciparum[3]. Unfortunately development of resistance in P. falciparum to various antimalarial drugs such as mefloquine, piperaquine, as well as artemisinin, has already been reported from Greater Mekong areas and several South-east Asian countries[4]. In India, malaria cases have reduced gradually over the years from over a million to around three hundred thousand between 2014 and 2018 while resistance to antimalarial drugs in P. falciparum has been developing at an alarming rate resulting in replacement of second line antimalarial drug combination of sulfadoxine and pyrimethamine with artemisinin derivatives in National Drug Policy[5], which warrants development of newer and more potent antimalarials. Continuous improvement of the drug discovery methods and high quality lead generation process are likely to deliver potential compounds with better therapeutic activity[6]. Artemisinin,a backbone of current antimalarial therapies, is a sesquiterpene lactone containing an endoperoxide linkage. This highly oxygenated sesquiterpene lactone peroxide is found to be a superior plasmocidal and blood schizontocidal agent, without adverse effects in patients[7]. Artemisinin was isolated by Chinese researchers in 1972 from Artemisia annua L (sweet or annual wormwood) and its structure elucidated in 1979[8]. The complex nature of the artemisinin molecule offers limited scope for random derivatization.1, 2, 4-trioxane system of artemisinin is mainly responsible for its antimalarial action[9]. The delicate nature of the 1, 2, 4-trioxane system beckons placid conditions for structural modifications[10]and majority of the derivatives have been prepared so far through modifications at the carbon positions 10 and 13 of the molecule.Prior to this, researchers have attempted transformation in the artemisinin nucleus using different microorganisms with moderate success, but their efforts led to further studies[11,12]. It has been observed that the C-10 non-acetal type artemisinin derivatives tend to be more hydrolytically stable, less toxic and possess a longer halflife[13,14]. Based on the theory of “ion trapping” in the parasitic food vacuole, artemisinin analogues containing basic amino functionality at the C-10 carbon were earlier synthesized with satisfactory results[15]. In search of water soluble artemisinin derivatives amino group containing compounds were synthesized and tested against
Plasmodium(P.) berghei and Plasmodium knowlesi with favourable results[16]. Mostly synthesized dimers are derivative ethers of dihydroartemisinin. Non-acetal C-10 olefinic dimers and saturated dimers synthesized from the natural trioxane artemisinin showed excellent results against the chloroquine-sensitive NF54 parasite strain[17]. Artemisinin dimers have been successfully synthesized by modification at the C-16 position of naturally occurring artemisitene via Michael addition and C-16-derived artemisinin dimers showed marked improvement in activity against the multidrug resistant K1 strain as compared to the standard artemisinin[18].A new series of hydrolytically stable, C-10 non-acetal, four-carbon atom linked trioxane dimers including phthalate and its derivatives were synthesized and evaluated against chloroquine-sensitive P.falciparum (NF 54) parasites. The compounds were found to be more potent than the parent compound artemisinin in in vitro studies[19].Hydroxylation of the unactivated carbons (C-4, C-5, C-6 and C-7) of the artemisinin was achieved by several workers. Recently a series of dimers were synthesized by using dinitroaliphatics as linkers[20].1,2,3-triazole family dimers have been shown to possess interesting biological properties[21]. In the present study we synthesized a series of artemisinin derivatives via Huisgen 1, 3-dipolar cyclo addition reaction, the most powerful click reaction to synthesize artemisinin dimers[22] and evaluated the synthesized dimers against the chloroquine-sensitive (3D7) and resistant (RKL2) P. falciparum strains aiming at obtaining artemisinin derivatives with potent antimalarial action with less toxicity. Further, we took PfATP6,the parasite orthologue of mammalian sarcoplasmic Ca2+ATPase(PfSERCA) that has been identified as a key molecular target of artemisinin[23] as target protein for molecular docking and structureactivity relationship to identify the residues and groups responsible for any increase/decrease in antimalarial activity of the compounds.
2. Materials and methods
All the chemicals for synthesis were procured from Merck(India)and for antimalarial evaluation from Sigma Aldrich(Germany). The chemicals were used as received.
2.1. Synthesis of compounds-chemistry
The work was carried out based on the strategies used by Saikia and his co-workers in which the synthetic scheme began with the synthesis of diazido aliphatic[24,25] and aromatic diazides[26](compounds from commercially available dihalides and also azido derivative of artemisinin (Figure 1).
Reaction of dibromoaliphatic compound 2a-c with sodium azide in dimethylformamide yielded the desired diazidoaliphatics 3a-c in 80%-86% isolated yields after 12 h. Artemisinin lactol 6 on treatment with sodium azide and bromotrimethylsilane in dichloromethane under inert atmosphere and at 25 ℃ for 12 h produced 10αazidoartemisinin 7 (GB-2)[27]. Artemisinin lactol was synthesized from artemisinin by sodium borohydride reduction in methanol at low temperature.

Figure 1. Scheme 1a: synthesis of aliphatic diazides; Scheme 1b: synthesis of aromatic diazides; Scheme 1c: synthesis of 10 -azidoartemisinin (GB2).

Figure 2. Synthesis of terminal alkynes of artemisinin viz. 8 and 9 (GB1).
The compounds with the alkyne linkage, viz. 8 and 9 (GB-1), were synthesized from artemisinin lactol by treatment with propargyl alcohol and homopropargyl alcohol respectively in presence of Aberlyst-15 in anhydrous dichloromethane at 25 ℃ (Figure 2).Huisgen 1,3-dipolar cycloaddition[28] between the synthesized aliphatic and aromatic diazides and two alkyne derivatives of artemisinin 8 and 9 (GB-1) were carried out in the presence of copper sulphate and sodium ascorbate in dichloromethane and water at 25 ℃ to obtain the desired artemisinin dimers with their yield percentages i.e. 10 (BS-8) (36%), 11a (BS-4) (38%), 12 (BS-3)(41%), 13 (BS-6) (40%), 14 (BS-7) (39%), 15 (BS-12) (40%),and 16 (BS-13) (40%) in isolated yield. In the case of 8 and 1,8-diazidoocatne, one artemisinin monomer derivative 11b (BS-5)(50%) could be isolated along with dimer 11a (BS-4). Reaction of 10α-artemisinin with 8 and 9 (GB-1) produced artemisininderived 1, 2, 4-trioxane dimers 17 (BS-4) (39%) and 18 (BS-15)(38%) with a 1, 2, 3-triazole ring system. The stereochemistry of the C-10 centers of azidoartemisinin and two alkyne derivatives of artemisinin remained identical for each dimer. It was found that cycloaddition of artemisinin-derived terminal alkynes with various diazides catalyzed by Cu (I) can be conducted at 25 ℃ leading exclusively to 4-substituted 1, 2, 3-triazoles (Figure 3). Initially,Nuclear Overhauser Effect Spectroscopy (NOESY) experiments were carried out to determine the stereochemistry of the compounds.However, NOE cross peaks were considered inconclusive because of the flexibility of the side chains of the compounds. Thereby the stereochemistry of the compounds was determined by synthesizing the corresponding MTPA (Mosher’s acid) ester. Attempts to synthesize MTPA-esters of these compounds were, however, futile and, therefore, the empirical method of Li- pase catalyzed kinetic resolution of alcohol by Kazl-auskas et al. was used[29].
Thus, a total of 15 compounds were synthesized and evaluated for antimalarial activity.
2.2. Molecular docking
2.2.1 Target identification
The PfATP6 is a 139 kD protein which is made up of 1 228 amino acids and has 51% resemblance to the mammalian SERCA protein,a target for the antimalarial drug artemisinin[30]. Accordingly,synthesized compounds were targeted against PfATP6 to check for their binding affinity and pattern considering artemisinin as a positive control ligand. The study provided crucial information regarding the antimalarial potential of the synthesized compounds.
Since the crystallographic structure of PfATP6 has not been developed, the modeled structure of PfATP6 (PDB ID: 1U5N)[31,32]was used for molecular docking purposes.
2.2.2 Ligand preparation
For docking, the ligands were prepared using ChemDraw Professional[33]. The prepared ligands in PDB files were finally used for docking. The “Prepare ligand” protocol of DS 4.5 was used to prepare the ligands which remove duplicate structures, standardizes the charges of common groups, calculates the ions and ionization of the ligand’s functional groups, generates isomers and tautomers, 2D-3D conversion, verify and optimize the structures, and other tasks established by user-defined parameters. The CHARMM force field was used to minimize the energy of the ligands.
2.2.3 Docking of receptor with ligands
PfATP6 was used as receptor molecule for docking to probe the binding free energy between the ligand and the receptor using fully functional trial version of Molegro Virtual Docker (MVD 2010.4.0) for Windows. MVD software is based on a new heuristic search algorithm that combines differential evolution with a cavity prediction algorithm[34]. The cavity prediction was also carried out using CASTP server (http://sts.bioe.uic.edu/castp/). For each compound ten runs were carried out and five poses of the ligands were selected. Default settings were applied to the rest of the docking parameters. The PDB structure file of ligands and protein complex generated from MVD software was analyzed by Discovery Studio 4.5 visualizer to identify specific interactions between the ligand and protein complex[35].

Figure 3. General synthetic scheme for artemisinin-derived dimers containing triazole moieties.
2.3. Antimalarial evaluation
2.3.1 Parasite cultivation
The chloroquine-sensitive (CQ sensitive) 3D7 and chloroquioneresistant (CQ resistant) RKL-2 strains of P. falciparum, procured from ICMR-National Institute of Malaria Research, New Delhi (India),were maintained in continuous culture following Trager and Jensen[36]with slight modifications. Briefly, the parasites were maintained at 6%haematocrit in RPMI-1640 medium supplemented with 25 mM HEPES,0.23% sodium bicarbonate, 0.2% D-glucose, 0.1% Gentamycin sulfate(50 mg/mL), 0.1 % Amphotericin B (250 μg/mL), and 15% human AB+ve serum at pH 7.2-7.4. The red blood cells used were O+ve and the culture plates were incubated at 37 ℃ in 5% CO2environment with regular medium change and parasitaemia assessment.
2.3.2 In-vitro antiplasmodial assay
For in vitro antiplasmodial evaluation of the synthesized compounds against CQ sensitive and CQ resistant strains of P. falciparum the method of Rieckmen et al.[37] with slight modifications was adopted.Compounds were dissolved in 1:200 dimethyl sulfoxide (DMSO)to get a stock solution of 5 mg/mL. Further dilutions of the stock solution were done with incomplete culture medium (without human AB+ serum). In a 96-well flat bottom microtitre plate required volumes of stock solution were charged in triplicate to which synchronized[38] 1% ring-stage parasitemia with 3 % hematocrit was added to get the desired dosages. The negative control (without compound) wells were made with only complete culture medium and 0.5 % DMSO. The reference drug artemisinin at its predetermined IC50dosage (inhibitory concentration to kill 50 % parasitaemia) was run concurrently as the positive control. The test plate was incubated for 40 h in a modular incubator at 37 ℃ and 5 % CO2environment.Thereafter, thin smears were prepared from each well, fixed with methanol, stained with 3% Giemsa and observed under microscope.A total of 100 asexual parasites (rings, trophozoites, and schizonts)were counted in each smear and scored as dead or alive based on parasite morphology. The antiplasmodial activity of the compound was measured in terms of dead parasites in test wells, which was calculated as a percentage of parasite growth in the control wells using the formula as follows:

In view of resource optimization all compounds were screened initially at 2 fixed dosages i.e. 0.1 μg/mL and 1.0 μg/mL in case of CQ sensitive strain; and 0.8 μg/mL and 8.0 μg/mL in case of CQ resistant strain. Two compounds (GB-1 and GB-2) showing promising in vitro antiplasmodial activity in initially screening were further evaluated in vitro at multiple dosages and their IC50values were obtained using the log concentration-response probit analysis program[39].
2.3.3 In-vivo antimalarial assay
Two compounds showing most promising in vitro antiplasmodial activity (GB-1 and GB-2) were evaluated in vivo against chloroquinesensitive rodent malaria parasite P. berghei in Swiss albino mice model. For animal experimentation, prior approval of Institutional Animal Ethical Committee of ICMR-Regional Medical Research Centre, Dibrugarh (India) was obtained (Approval no. RMRC/Dib/IAEC (Animal)/Certificate of approval, dated20/09/2013). The rodent malaria parasite P. berghei was obtained from CSIR-Central Drug Research Institute, Lucknow (India) and maintained in mice through serial passage. In-vivo testing was carried out using Peters’4-day suppressive test[40]. Chloroquine diphosphate at 5 mg/kg bw dosage and artemisinin at 50 mg/kg bw dosage were used as reference drugs. The test compounds were evaluated at two dosages i.e. 5 mg/kg bw and 50 mg/kg bw. Four to six weeks old healthy mice weighing about 20-25 g were selected and divided into treated(with test compounds), untreated (negative control) and positive controls (treated with chloroquine and artemisinin) with three mice in each group/dosage. On the first day (Day 0) each mice was inoculated intraperitonially with 18.18 μL of 1× 107parasitaemia of P. berghei. After 2 h of inoculation, 0.2 mL of the test compounds/chloroquine/ artemisinin (dissolved in 1 % DMSO) was administered intraperitonially and to the negative group 0.2 mL of 1% DMSO was administered. Daily administration of test compound/chloroquine/artemisinin/DMSO solution continued up to Day 3 and on Day 4 thin smears were prepared in duplicates from the tail blood of each mouse. The smears were air dried, fixed with methanol, stained with 3% Giemsa and observed under microscope. Percent parasitemia was determined by scanning 5 000 erythrocytes in random fields.Parasitaemia suppression in each treatment was calculated with reference to control by using the formula:

For each group the mean survival time of the treated mice was determined.
3. Results
For in vitro antiplasmodial activity detection, initially compounds were evaluated at two fixed dosages viz. 0.1 μg/mL (equivalent to IC50value of the reference drug artemisinin) and 1.0 μg/mL(10 times of IC50value of the reference drug artemisinin) against CQ sensitive 3D7 strain of P. falciparum. The corresponding test dosages of compounds against CQ resistant RKL-2 strain were 0.8 μg/mL and 8.0 μg/mL similarly. Two compounds viz. GB-1 and GB-2 exhibited promising activity that was almost equal to artemisinin at lower dosage (Table 1).
These 2 compounds were further investigated and their IC50values were determined (Table 2). Between the two compounds, GB-2 recorded superior antiplasmodial activity with IC50value of 0.066 μg/mL compared with the IC50of GB-1 (0.215 μg/mL) against 3D7 strain and was also superior to artemisinin (IC50: 0.1 μg/mL). Also, GB-2 recorded superior activity (IC50: 0.865 μg/mL) against RKL-2 strain compared to GB-1 (IC50: 8.0 μg/mL) and the acitivity was equivalent to artemisinin(IC50: 0.8 μg/mL).
Both the compounds, especially GB2, effectively suppressed parasitaemia levels of P. berghei in vivo in mice (Table 3). GB-2 achieved 55.5% parasitaemia suppression at the lower dosage of 5.0 mg/kg bw. However, its activity at similar dosage was inferior to the reference compound chloroquine and at par with artemisinin.In comparison, GB-1 was just not effective at the lower dosage but at higher dosage of 50.0 mg/kg bw was at par with GB-2 and artemisinin. The mean survival time of mice treated with GB-2 at higher dosages was (13.3 ± 8.1) days compared to (15.3 ± 9.8) days of mice treated with GB-1 (Table 3) and was much less than the mean survival of mice treated with chloroquine (30.0 ± 0.0) days.Interestingly mice treated with the higher dosage (50.0 mg/kg bw)of GB-2 and GB-1 survived for less number of days as compared to mice treated with the lower dosage (5.0 mg/kg bw) suggesting probable toxicity of the compounds to mice at the higher dosage.
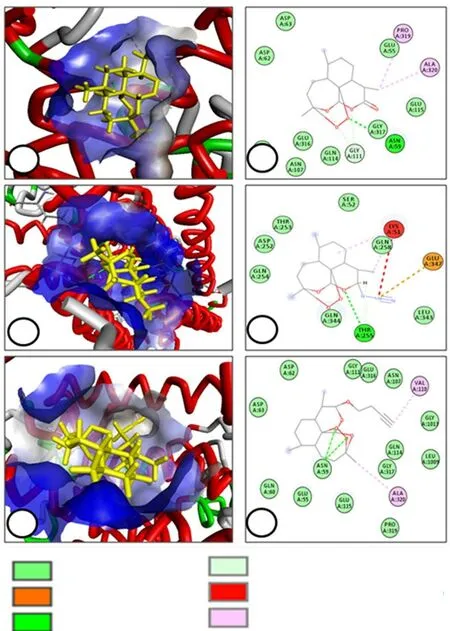
Figure 4. Molecular docking interactions of PfATP6 protein with artemisinin (A & B), GB-2 (C & D) and GB1 (E & F).

Table 1. Initial in vitro antiplasmodial activity of the synthesized compounds against Plasmodium falciparum (mean±SD).
Using molecular docking and visualization, an attempt was made to analyze the different binding interaction of GB-1 and GB-2 compounds with PfATP6 molecule and postulate structure-activity relationship. Left panel of the Figure 4 shows accommodation of ligands during docking in the active site of target protein and right panel indicates different types of interactions with the specific amino acid residues. Artemisinin was used as positive reference ligand. Molegro Virtual Docker (MVD 2010.4.0) software used for docking study provides H-bond energy values as negative, zero or positive and the low negative score suggests stable protein-ligand interaction. Compounds GB-1 and GB-2 had higher binding energies as compared to that of the reference artemisinin (Table 4).
The spectroscopic data for the most promising compound GB-2 is as follows:1H NMR(300 MHz, CDCl3) d 5.39(1H, s, H-12), 4.61(1H, d, J = 10.2 Hz, H-10), 2.42(1H, m), 2.37(1H, m), 2.04(1H, m), 1.45(3H, s, 3-CH3),0.97(3H, d, J = 5.9 Hz, 9-CH3), 0.92(3H, d, J = 7.1 Hz, 6-CH3)ppm;13C NMR(75 MHz, CDCl3) d 104.5, 91.7, 87.7, 80.0, 51.6, 45.2,37.3, 36.1, 34.0, 32.5, 25.9, 24.7, 21.7, 20.2, 12.9 ppm.

Table 2. IC50 value of the two promising compounds GB-1 and GB-2 against Plasmodium falciparum (mean±SD).
The1H NMR spectra at 5.39 is indicative of the hydrogen present in the 7thposition of the structure and its distance from the other peak data indicates its high shielding due to the presence of the oxygen atoms at the adjacent side. The peak data at 4.61 shows the presence of the hydrogen next to the nitrogen groups in the position 9. The peak data at 0.97 and 0.92 signifies the presence of the hydrogens in the methyl group at positions 19 and 21. The data at 1.45 is that of the methyl hydrogens present in the position 17. In13C NMR the farthest data peak at 104.5 is the carbon peak at position 13.

Table 3. In vivo antimalarial activity of compounds GB-1 and GB-2 against Plasmodium berghei in Swiss albino mice.

Table 4. Docking score of various ligands with the receptor molecule PfATP6.
4. Discussion
In the present work a total of 15 compounds belonging to a novel series of artemisinin derivatives containing 1,2,3-triazole moiety were synthesized and evaluated for antiplasmodial activity in vitro against both chloroquine-sensitive and resistant P. falciparum strains of human malaria parasite and in vivo against P. berghei rodent malaria parasite. Two compounds i.e. GB-1 and GB-2 showed the most promising antiplasmodial activity, especially GB-2 which recorded superior in vitro activity than that of the standard artemisinin. Of these, GB-2 is a 10 α-azido artemisinin and GB-1 is a terminal alkyne, linked by two C-atoms. This suggests that the modification of the parent artemisinin compound at position 10 resulted in good antiplasmodial activity. Molecular docking was performed to corroborate the antiplasmodial activity of GB-1 and GB-2 compounds. Structure-activity relationship revealed that increase/decrease in the carbon chain length between different groups did not alter the antiplasmodial activity of the compounds appreciably.It was noted that artemisinin interacts with the Gly317 residue of PfATP6 via conventional hydrogen bonds. The 2D structure revealed that the interaction took place between the endoperoxide oxygens of artemisinin and the residue Asn59. Lipophilic alkyl interactions were seen with the methyl group at position 13 and the Pro319 and Ala320 residues. A similar connection was observed between the GB-1 and Asn59 which, however, was due to the bond between the endoperoxide oxygens. GB-1 showed lipophilic interactions via alkyl bonds between the alkyne side chain at position10 and carbon of position 3 with that of Val110 and Ala320 respectively.This indicated that besides working by the normal mechanism of decomposition of the endoperoxide linkage to release carbon centred free radicals, the bonding of GB-1 with Asn59 intensified its activity. GB-2 showed conventional hydrogen bonding between the oxygen at the tenth position and hydrogen at ninth position with the residue Thr255. This hydrogen bond could be the reason for the added increase in activity of the GB-2 compound in comparison with that of artemisinin and GB-1. An unfavourable bond was also observed between the second nitrogen of the triazyne group at position 9 of the artemisinin nucleus with Lys51. A strong attractive charge was observed with the same nitrogen with that of Glu347 residue. The docking results suggest that the residue Asn59 is a common site of action for artemisinin and GB-1. Although the binding atoms are different for artemisinin and GB-1 yet both show favorable interactions with Asn59. Further, GB-1 did not show any interaction with Gly317 as was seen in case of artemisinin. GB-2 on the other hand did not interact with either Asn 59 or Gly317 residue.However, its strong bonding with Thr255 residue is likely to be the main reason for its excellent antimalarial activity despite showing an unfavourable link with Lys51. Further research is required to explore the interaction of GB-2 with Thr255 to analyse its pharmacophoric property. In general, compounds with a single triazole moiety have recorded better activity than bis triazole compounds which can be attributed to the free azide group lacking in the other compounds.It is concluded that the compound GB-2 may be utilized as a lead molecule for further chemical modifications to improve its potential as an antimalarial agent and further synthesis of compounds with similar analogy could be a key to discovering newer and more potent antimalarial drug candidates.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Acknowledgements
We are thankful to the Director, ICMR-National Institute of Malaria Research, New Delhi, India and Director, CSIR-Central Drug Research Institute, Lucknow (UP), India for providing human malaria parasites and rodent malaria parasites respectively used in this study. Physical facilities and financial support as an intramural activity provided by Director, ICMR- Regional Medical Research Centre, Dibrugarh (Assam), India for the study is gratefully acknowledged. We are grateful to the members of Department of Natural Products Chemistry Division, CSIR-NEIST, Jorhat (Assam),India and members of Malariology Division, ICMR- Regional Medical Research Centre, Dibrugarh (Assam), India for helping in various ways during the study.
 Asian Pacific Journal of Tropical Medicine2019年5期
Asian Pacific Journal of Tropical Medicine2019年5期
- Asian Pacific Journal of Tropical Medicine的其它文章
- Parsonage-Turner syndrome following chikungunya virus infection:A case report
- Seroprevalence of Cryptosporidium and risks of cryptosporidiosis in residents of Sothern Egypt: A cross-sectional study
- Evaluation of phytochemical properties and larvicidal activities of Cynodon dactylon, Clerodendrum viscosum, Spilanthes acmella and Terminalia chebula against Aedes aegypti
- Phylogeny of Culex theileri virus flavivirus in Spain, Myanmar, Portugal and Turkey
- Climate change and potential distribution of zoonotic cutaneous leishmaniasis in Central Iran: Horizon 2030 and 2050