
竹节参HPLC指纹图谱的建立及7种成分测定
2019-06-03 08:05伍红年谭诗涵王元清雷雅婷刘瑞连严建业
中成药 2019年5期
伍红年谭诗涵王元清雷雅婷刘瑞连严建业∗
(1.湖南中医药大学药学院,湖南 长沙 410208;2.湖南中医药大学科技创新中心,湖南 长沙 410208;3.中南林业科技大学生命科学与技术学院生物技术与工程实验室,湖南 长沙 410004;4.湖南省中医药研究院,湖南 长沙 410006)
竹节参又名白三七、竹节七、竹节人参等,系五加科人参属植物竹节参Panax japonicusC.A.Mey 的干燥根茎,收载于2015年版 《中国药典》一部,具有散瘀止血、消肿止痛、祛痰止咳、补虚强壮之功效[1]。现代研究表明,竹节参具有保护心肌功能、促进免疫、抗肿瘤、抗惊厥、抗应激和抗氧化等药理作用;已报道的化学成分有皂苷类、糖类、多炔类、氨基酸、挥发油无机元素及核苷类[2-3]。
然迄今为止,关于竹节参相关质量控制研究主要集中于皂苷[4]、多糖[5]及挥发性[6]成分,尚未见对核苷类成分质量控制相关的研究报道。核苷类成分具有广泛的生理活性,是生物细胞维持生命活动的基本组成元素,具有抗肿瘤、抗病毒、免疫调节、改善细胞代谢、镇静中枢神经、抗血小板凝集、抗心律失常和抗惊厥等作用[7],与竹节参药效具有一定关联性;吴兵等[3]在对竹节参进行化学成分研究中,首次从中分离得8种核苷类成分;张元杰等[8]认为核苷可能是补益中药的共同物质基础,对三七、人参等6种常用的补益药进行研究,结果表明均含有核苷类。因此核苷可作为竹节参药材的质量评价指标之一。
指纹图谱作为1种多组分复杂样品的有效质量控制方法,能够反映出待测样品的整体性、特征性[9],结合同时测定多种成分的含有量,被广泛应用于中药材和中成药的质量研究中[10-11]。本研究采用HPLC 法建立了竹节参核苷类成分的指纹图谱,结合聚类与主成分分析进行分类分析,并测定其中7种主要成分的含有量,以期为竹节参质量控制提供依据。
1 材料
Waters e2695 型高效液相仪、Waters 2489 UV检测器、Empower 3 色谱数据工作站(美国Waters公司);LE204E/02 型电子天平(上海梅特勒-托利多有限公司);Option R7 utra AN 超纯水系统(英国 Elga Lab Waters 公司);eppendorf 5424R 高速冷冻离心机;KQ-5200DV 型数控超声波清洗仪(昆山市超声仪器有限公司)。乙腈色谱纯;水为超纯水;其他试剂均为分析纯。次黄嘌呤(批号wkq16091402)、尿苷(批号 wkq16083105)、脱氧尿苷(批号 wkq17011101)、肌苷(批号 wkq16090505)、腺苷(批号wkq16092103)、胸腺嘧啶核苷(批号wkq16090903)、鸟苷(批号 wkq16090703)均购买于四川省维克奇生物科技有限公司,含有量均≥98%;10 批不同产地竹节参样品,经湖南中医药大学龚力民副教授鉴定为五加科人参属植物竹节参Panax japonicusC.A.Mey 的干燥根茎,见表1。

表1 样品信息Tab.1 Information of samples
2 方法与结果
2.1 色谱条件 Venusil MP C18色谱柱(250 mm×4.6 mm,5 μm);流动相为乙腈(A)-0.01% 甲酸水(B),梯度洗脱(0~15 min,0% A;15~20 min,0%~1%A;20~25 min,1%~3%A;25~55 min,3% A;55~60 min,0% A);体积流量0.8 mL/min;检测波长 254 nm;柱温 30 ℃;进样量 10 μL。
2.2 溶液制备
2.2.1 混合对照品溶液制备 分别取次黄嘌呤、尿苷、脱氧尿苷、鸟苷、肌苷、腺苷和胸腺嘧啶核苷对照品适量,精密称定,加超纯水制成每1 mL分别含次黄嘌呤 5.60 μg、尿苷 77.59 μg、脱氧尿苷 5.52 μg、肌苷 21.79 μg、鸟苷 80.00 μg、腺苷36.41 μg、胸腺嘧啶核苷 14.00 μg 的溶液,作为混合对照品溶液。
2.2.2 供试品溶液制备 取样品粉末(过40 目筛)约 1.0 g,精密称定,置于具塞锥形瓶中,加入 0.1% 氨水 10 mL,称定质量,超声提取(40 kHz、250 W)90 min,放冷,再称定质量,用0.1% 氨水补足减失质量,13 000 r/min 离心10 min,0.45 μm 微孔滤膜过滤,即得。
2.2.3 空白溶液制备 0.1%氨水溶液。过滤,进样,记录色谱图。见图1,空白无干扰。
2.3 HPLC 指纹图谱建立
2.3.1 精密度试验 取同一供试品(S10),按“2.2.2”项下方法制备供试品溶液,在 “2.1”项色谱条件下连续进样6 次,以尿苷为参照峰,测得16 个共有峰相对保留时间和相对峰面积RSD 介于0.04%~0.30% 及 0.87%~2.99% 之间,均小于3.0%,表明仪器精密度良好。

图1 各成分HPLC 色谱图Fig.1 HPLC chromatograms of various constituents
2.3.2 重复性试验 取同一供试品(S10),按“2.2.2”项下方法制备供试品溶液 6 份,在“2.1”项色谱条件下进样测定,测得各共有峰相对保留时间和相对峰面积RSD 介于0.05%~0.61%及0.86%~2.71%之间,表明该方法重复性良好。
2.3.3 稳定性试验 取同一供试品(S10),按“2.2.2”项下方法制备供试品溶液,于 0、2、4、6、8、10、12 h 在 “2.1”项色谱条件下进样测定,测得各共有峰相对保留时间和相对峰面积RSD 介于 0.10%~0.89% 及 2.00%~2.88% 之间,表明供试品溶液在12 h 内稳定性良好。
2.3.4 指纹图谱建立 吸取S1~S10 供试品溶液及混合对照品溶液各10 μL,进样后得到10 批样品的HPLC 指纹图谱,见图2。将所得图谱导入“中药色谱指纹图谱相似度评价系统2004A”进行分析,获得对照指纹图谱R,共得到16 个共有峰,将所得对照指纹图谱与对照品比对可知,确定了其中7 个有峰,分别为次黄嘌呤(5 号峰)、尿苷(8 号峰)、脱氧尿苷(10 号峰)、肌苷(11 号峰)、鸟苷(12 号峰)、腺苷(13 号峰)和胸腺嘧啶核苷(15 号峰)。各批样品中尿苷的峰面积较大,分离度较好,因此确定其为参照峰S 计算各共有峰的相对峰面积和相对保留时间,结果见表2~3。

图2 10 批样品HPLC 指纹图谱Fig.2 HPLC fingerprints of ten batches of samples
2.4 相似度分析 将10 批竹节参色谱图与对照指纹图谱相比较,各批次样品的相似度在0.723~0.978,表明各批次竹节参样品的质量相对稳定,从图2 可以看出不同产地竹节参的化学成分基本一致,各成分在量上存在差异。表4 表明,样品 S2与S3 相似度相对较低,而其他产地样品相似度则较高。
2.5 聚类分析 聚类分析是通过将观察对象依据某些特征加以归类,从而建立样本与样本之间的相似关系或亲疏关系的一种探索性数据分析手段,目前已广泛应用于中药鉴别、质量评价和品种分类等方面[12]。采用 SPSS 19.0 软件,以 10 个不同产地的竹节参药材的16 个共有峰的峰面积为变量,选用组间联接法,Euclidean 距离为测度,进行聚类,结果见图3。表明,当类间距离范围为 15~25 时,竹节参药材可分为3 类,其中S4(四川大凉山)、S5(湖南石门)、S7(贵州贵阳)、S8(湖北神农架)、S9 与 S10(湖北恩施)为Ⅰ类;S1(云南丽江)与S6(江西吉安)为Ⅱ类;S2(四川乐山)与S3(四川雅安)为Ⅲ类。结果表明,竹节参药材的指纹图谱与其地理位置,外界环境具有一定的相关性,但不绝对相关。

表2 10 批样品共有峰相对保留时间Tab.2 Relative retention time of common peaks of ten batches of samples

表3 10 批样品共有峰相对峰面积Tab.3 Relative peak areas of common peaks of ten batches of samples

表4 10 批样品相似度Tab.4 Similarities of ten batches of samples
2.6 主成分分析 为进一步探讨不同产地竹节参之间的差异,在聚类分析的基础上,对指纹图谱数据进行主成分分析。以10 个不同产地的竹节参药材的16 个共有峰的峰面积导入SPSS 19.0,对共有峰峰面积进行标准化处理,进行主成分分析。主成分个数提取原则为主成分的特征值大于1 的前m个主成分,表5 表明,前4 个主成分的特征值大于1,即 5.652、3.996、2.004、1.459,对方差的贡献 率 分 别 为 35.328%、24.976%、12.525%、9.117%,累计贡献率为 81.946%。表 7 表明,竹节参的多个成分可以简化为4 个主成分;图4 表明前4 个成分较陡,剩余的其他成分之间比较平缓。因前3 个主成分的特征值相对较大,故取前3 个主成分的得分作不同产地竹节参药材主成分三维分布图,见图5。表明,S4、S5、S7、S8 和 S9 与 S10为Ⅰ类;S1 与 S6 为Ⅱ类;S2 与 S3 为Ⅲ类;其结果与聚类分析一致。

图3 10 批样品聚类树状图Fig.3 Dendrogram of ten batches of samples

表5 主成分的初始特征值和贡献率Tab.5 Initial eigenvalue and contribution rate of princilal component

图4 各样品主成分分析碎石图Fig.4 PCA scree plot of various samples
2.7 含有量测定
2.7.1 线性关系考察 取混合溶液 1、4、8、12、16、20 μL,在 “2.2.1”项色谱条件下进样,以色谱峰峰面积为纵坐标(Y),进样量为横坐标(X)进行回归,结果见表 6。
2.7.2 精密度试验 取 “2.2.1”混合对照品溶液,在 “2.1”项色谱条件下连续进样 6 次,测得次黄嘌呤、尿苷、脱氧尿苷、肌苷、鸟苷、腺苷和胸腺嘧啶核苷峰面积RSD 分别为0.41%、0.39%、0.42% 、0.37%、0.39% 、0.36% 、0.48% ,表 明仪器精密度良好。

图5 不同产地样品主成分3D 得分图Fig.5 3D PCA score of samples from different growing areas

表6 各成分线性关系Tab.6 Linear relationships of various constituents
2.7.3 重复性试验 取同一供试品(S10)6 份,按 “2.2.2”项下方法制备供试品溶液,在 “2.1”项色谱条件下进样测定,测得次黄嘌呤、尿苷、脱氧尿苷、肌苷、鸟苷、腺苷和胸腺嘧啶核苷峰面积的 RSD 分别为 2.13%、2.26%、2.77%、2.88%、2.80%、2.41%、2.47%,表明该方法重复性良好。
2.7.4 稳定性试验 取同一供试品(S10),在“2.1”项色谱条件下,于 0、2、4、6、8、10、12 h 进样测定,测得次黄嘌呤、尿苷、脱氧尿苷、肌苷、鸟苷、腺苷和胸腺嘧啶核苷峰面积RSD 分别 为 2.28%、2.44%、2.92%、2.20%、2.81%、2.43%、2.87%,表明供试品溶液在12 h 内稳定性良好。
2.7.5 加样回收率试验 取 “2.3.3”项下已测知核苷含有量的样品6 份,每份约0.5 g,精密称定,精密加入次黄嘌呤、尿苷、脱氧尿苷、肌苷、鸟苷、腺苷和胸腺嘧啶核苷各对照品适量,按“2.2.2”项下方法制备供试品溶液,在 “2.1”项色谱条件下进样测定。结果次黄嘌呤、尿苷、脱氧尿苷、肌苷、鸟苷、腺苷和胸腺嘧啶核苷的回收率分别为 98.66%(RSD = 2.69%)、96.90%(RSD =2.92%)、93.34%(RSD=2.97%)、94.86%(RSD=2.82%)、98.60%(RSD=2.73%)、92.74%(RSD=2.66%)、97.95%(RSD=2.31%)。
2.7.6 样品含有量测定 吸取 “2.2.1”、“2.2.2”项下对照品溶液和S1~S10 供试品溶液各10 μL,在 “2.1”项色谱条件下进样,按外标法计算含有量,结果见表7。
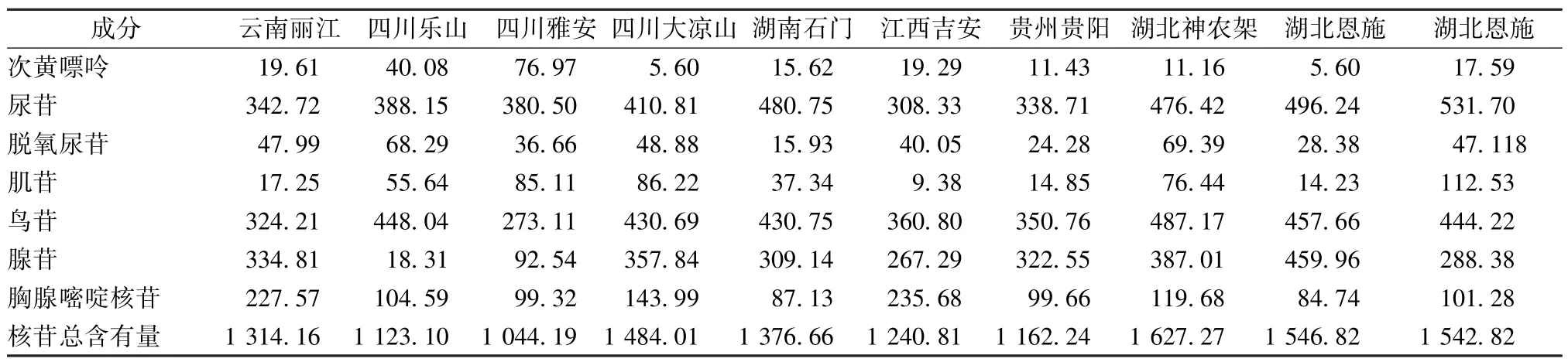
表7 各成分含有量测定结果(μg/g, n=3)Tab.7 Results of content determination of various constituents(μg/g, n=3)
2.8P值分析 为了探究各产地之间的质量波动,引用1 个易于计算和可见的参数(P值),对不同产地的药竹节参药材质量进行评价,计算公式为其中Ci表示给定化合物的测定浓度,而Ci表示10 个不同产地竹节参的平均浓度。P值越接近1,批间的一致性越好。一般来说,在0.75~1.25 的范围内的值是可以接受的[13],见图 6,从中观察到3 个异常值,即峰5(次黄嘌呤)、峰10(脱氧尿苷)和峰11(肌苷)。异常值是导致竹节参样品质量波动的重要因素。表明次黄嘌呤、脱氧尿苷、肌苷在样品中存在较大的波动,对竹节参的质量有较大影响。
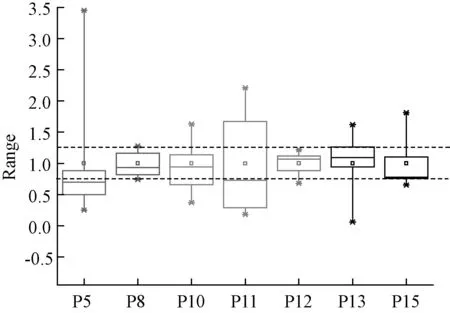
图6 不同产地样品7种成分Box-chart 图Fig.6 Box chart of seven compounds of samples from different growing areas
3 讨论
3.1 条件优化 由于核苷类成分极性较大,且有些核苷类成分(如次黄嘌呤、鸟苷)在水中的溶解度较差,在碱性溶剂中溶解度较好[14-15],所以考察了不同极性与不同pH 的提取溶剂(超纯水、10% 甲醇水、20% 甲醇水、10% 乙醇水、0.1% 氨水、1%氨水、1%醋酸水、0.1%醋酸水);不同提取方式(超声、回流);不同提取时间(30、45、60、90、120 min)对提取效率的影响。结果发现,采用0.1%氨水提取效率优于其他,且结果稳定;超声提取和回流提取的效果无明显差异,但超声提取法处理简单,重复性好;提取时间90 min 较适宜;因此选择0.1%氨水超声提取90 min 为供试品溶液的制备方法。
鉴于核苷类成分为偏碱性的水溶性成分,本实验考察了各种流动相后发现,使用乙腈-0.01% 甲酸水流动相时分离度较好,峰形对称;比较柱温对峰形的影响,考察了25、30、35 ℃ 3种不同温度,发现随着柱温的升高与降低,对其峰形都有较大影响,其中30 ℃下影响最小,选为本实验柱温;在其他色谱条件一致的情况下,分别对0.5、0.8、1 mL/min 3种体积流量进行考察,结果表明0.8 mL/min最好。
3.2 结果分析 本研究选取不同产地竹节参药材作为研究对象,建立了HPLC 指纹图谱及7种指标性成分含有量测定的方法,完成了在同一色谱图下对竹节参核苷类成分定性及定量分析的双重目的。由10 批样品指纹图谱可知,各共有峰相对保留时间差异较小,而相对峰面积差异则较大,表明不同产地竹节参样品中化学成分在量上存在较大差异。综合相似度评价、聚类分析和 PCA 分析结果,将四川大凉山、湖南石门、贵州贵阳、湖北神农架和恩施聚为Ⅰ类;云南丽江、江西吉安聚为Ⅱ类;四川乐山和雅安聚为Ⅲ类。为了进一步了解不同产地竹节参药材中核苷类成分的含有量的差异来源,对竹节参样品多指标性成分进行定量分析,并结合1个易于计算和可见的参数P进行分析,得到次黄嘌呤、脱氧尿苷、肌苷在样品中存在较大的波动,对竹节参的质量有较大影响。本实验建立了竹节参核苷类指纹图谱研究方法,该方法简便、重复性好,能够较好地识别竹节参中核苷类化学成分,监测指标性成分含有量的同时,又对样品整体进行了表征,对评价竹节参的质量具有参考意义。
猜你喜欢
临床肝胆病杂志(2022年6期)2022-11-25
中国药业(2022年15期)2022-08-09
河南农业·综合版(2022年2期)2022-03-18
昆明医科大学学报(2021年2期)2021-03-29
小哥白尼(趣味科学)(2021年11期)2021-02-28
小天使·一年级语数英综合(2020年10期)2020-12-16
山西中医药大学学报(2020年2期)2020-06-06
分析化学(2018年12期)2018-01-22
儿童时代·快乐苗苗(2016年2期)2016-10-22
Asian Journal of Urology(2015年3期)2015-12-16
