
不吸水链霉菌中沉默基因簇的激活和产物结构鉴定
2019-06-01 07:03:38钟磊孙青枝李博尹楚洁赵寿媛夏焕章
中国抗生素杂志 2019年5期
钟磊 孙青枝 李博 尹楚洁 赵寿媛 夏焕章
(沈阳药科大学生命科学与生物制药学院,沈阳 110016)
董德鑫等[1]1978年在广西梧州地区土壤中分离得到一株不吸水链霉菌的亚种(S.ahygroscopicus)。该菌产生的次级代谢产物对多种植物病原真菌有较强的抑制和杀死作用,是防治植物病害较好的生物农药[2]。陈文君等[3]在该菌的发酵液中分离得到了A1、A2、B和C 4个组分,其中A1、A2分别为多烯大环内脂类四霉素A(tetramycin)和四霉素B;B为肽类抗生素白诺氏菌素(albonursin);C为含氮杂环类芳香族衍生物茴香霉素(anisomycin)。任君等[4]在不吸水链霉菌梧州亚种的突变株S91(S.ahygroscopicus S91, 菌种保藏号CGMCC4.7082)发酵液中分离得到了制霉菌素及丰加霉素,通过生物信息学分析及基因功能研究发现并确定了其生物合成基因簇和部分合成基因的功能。通过对吸水链霉菌的亚种基因组进行分析,该菌种除产生这些抗生素外还具有产生谷氏菌素等其他抗生素的能力,但目前在该菌种的发酵产物中并未分离得到这些抗生素。
谷氏菌素(又名星霉素或无穗霉素)是一种胞嘧啶核苷肽类抗生素,呈弱碱性,易溶于水,不溶于乙酸乙酯、氯仿、甲醇、乙醇等一般有机溶剂,室温下不稳定,纯品易被氧化变黄。谷氏菌素最早是由日本科学家从谷氏链霉菌中分离得到的,并在1968年确定了其化学结构,其主要由核苷和肽基两部分组成[5]。近年,通过生物信息学分析、构建基因阻断突变株、异源表达的方法,初步探索了谷氏菌素的生物合成途径及相关基因的功能[6-7]。
谷氏菌素的生物合成是由多步酶催化反应,酶催化反应受分级调控。其生物合成的前体主要有UDP-葡萄糖、胞嘧啶、甘氨酸和三磷酸甘油酸。在谷氏菌素的生物合成基因簇中,gouA、gouB、gouF和gouH参与了谷氏菌素核苷骨架的形成[6-7]。
王锦[8]前期构建了同时阻断四霉素、制霉菌素和丰加霉素生物合成的阻断菌株不吸水链霉菌ΔttmS1 ΔnysB ΔtymH,该菌株发酵产物中仍然产生抗真菌抗生素。本课题通过分离纯化对该阻断菌株的发酵产物进行鉴定,证明该菌株能够产生云南霉素和谷氏菌素。
1 材料与方法
1.1 菌种
不吸水链霉菌ΔttmS1 ΔnysB ΔtymH:四霉素、制霉菌素、丰加霉素的生物合成阻断菌株,本实验室构建并保藏。
黑曲霉:生物活性检定菌,本实验室保藏。
1.2 培养基
1.2.1 斜面培养基(g/L)
可溶性淀粉20.0,牛肉膏1.0,K2HPO4·3H2O 0.5,MgSO4·7H2O 0.5,NaCl 0.5,KNO30.1,FeSO4·7H2O 0.01,琼脂15.0,pH7.2,121℃灭菌30min。
1.2.2 种子培养基(g/L)
葡萄糖20.0,酵母粉6.0,蛋白胨6.0,NaCl 10.0,pH7.2,121℃灭菌30min。
1.2.3 发酵培养基(g/L)
玉米淀粉8.0,玉米粉20.0,葡萄糖30.0,黄豆饼粉30.0,(NH4)2SO42.5,NaCl 0.2,K2HPO4·3H2O 0.2,MgSO4·7H2O 0.2,FeSO4·7H2O 0.2,轻质碳酸钙5.0,pH7.2,121℃灭菌30min。
1.2.4 沙氏培养基(g/L)
用于培养黑曲霉。蛋白胨10.0,葡萄糖40.0,琼脂12.0,pH自然,115℃灭菌30min。
1.3 主要试剂和仪器
琼脂(青岛海燕琼胶有限公司),三氯乙酸(分析纯,山东西亚化学工业有限公司),无水乙醇(天津市大茂化学试剂厂),制霉菌素标准品(中国食品药品检定研究院),DM-2树脂(山东鲁抗立科),732树脂(山东鲁抗立科)。
LC-10AT型高效液相色谱仪(日本Shimadzu)SPD-10A型高效液相紫外检测器(日本Shimadzu),GL-21M高速冷冻离心机(湖南湘仪实验室仪器开发有限公司),SC-3612型低速离心机(湖南湘仪实验室仪器开发有限公司),N-1001旋转蒸发仪(上海爱朗仪器有限公司),立式压力蒸汽灭菌锅(上海博讯实业有限公司医疗设备厂),离子阱质谱仪(美国Thermo Electron),布鲁克核磁共振仪(德国Bruker科学仪器公司)。
1.4 发酵培养
发酵液是经斜面孢子培养、种子培养、发酵培养的步骤进行的:将不吸水链霉菌生物合成阻断株S.ahygroscopicus ΔttmS1 ΔnysB ΔtymH菌种传至合V斜面培养基上,培养6~7d,待孢子变灰后挖菌块接种到种子培养基,28℃、220r/min摇床振摇24h获得种子液,按10%的接种量转接到发酵培养基中,28℃,220r/min摇床振摇96h。
1.5 发酵液预处理
用草酸将发酵上清液调pH3.0,煮沸10min,4000r/min,离心10min,弃沉淀,收集上清,备用。
1.6 样品抑制真菌实验(纸片法)
向检定盘中加入200mL加热融化的培养基,使其在检定盘底部均匀摊布,凝固后作为底层。另取100mL 沙氏培养基加热融化后,放冷置48~50℃,加入检定菌,摇匀倒入铺有底层培养基的检定盘中,作为菌层,培养基凝固后,将加有样品的纸片贴于菌层上,在28℃培养箱中培养12h,观察抑菌情况,将有抑菌活性样品收集,备用。
2 结果
2.1 发酵结果
发酵液经斜面孢子培养、种子培养、发酵培养之后,收集上清。以制霉菌素为对照品,对原始发酵液进行生物效价测定,其效价为159.57U/mg,纯度为2.4%(表1)。
2.2 发酵液的大孔吸附树脂DM-2脱色处理
发酵液经草酸处理后,黏度变小,变澄清。这是由于在酸性环境及加热情况下杂蛋白、多糖等高分子杂质变性形成沉淀,纯度提高到3.37%。
考察pH值对树脂吸附效果的影响。取60mL预处理发酵液,均分到6个250mL锥形瓶中,每瓶10mL,分别用1mol/L HCl和1mol/L NaOH调pH为1、3、5、7、9和11,向其中加入2g DM-2大孔吸附树脂,置于28℃,220r/min摇床振摇4h,静置30min。如图1所示,发现在pH1、3和5时脱色效率较高。通过生物效价测定经树脂吸附后洗脱液的活性损失率(图2),pH1时活性物质损失最少,所以在后续试验中采用pH1对发酵液进行脱色处理。pH1时每10mL发酵液中脱色树脂的加入量为1.0g。
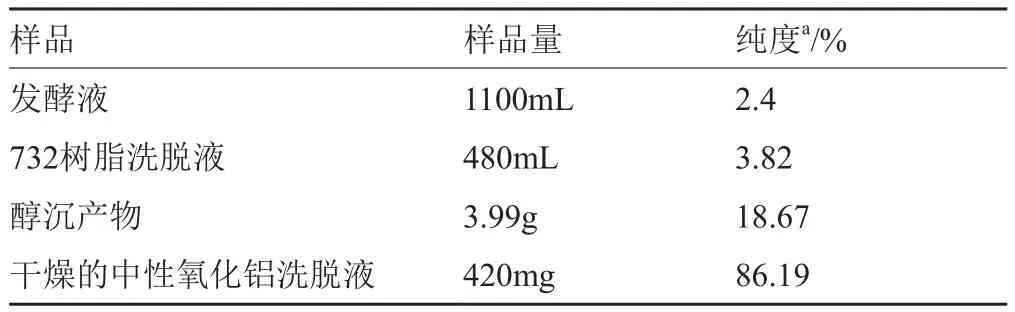
表1 各纯化过程中样品量和产物纯度变化Tab.1 Changes in sample amount and product purity during each purification process

图1 在不同pH条件下DM-2树脂脱色前后对比Fig.1 Comparison of decolorization of DM-2 resin at different pH conditions
2.3 发酵液的732离子交换树脂处理
本实验采用NH3·H2O进行解吸。探索了树脂静态吸附动力学曲线、穿透曲线、动态解吸曲线(图3~5)。
2.4 醇沉实验
将732树脂的洗脱液合并,减压浓缩后,用36%乙酸调pH至4.5,加8BV 95%乙醇置于4℃冰箱过夜,然后4℃,4000r/min,离心10min,取沉淀挥干,得到3.99g褐色粉末即活性组分的盐。经醇沉后纯度提高到18.67%,效价达到1231.12U/mg。如表1产物纯度得到了提高。
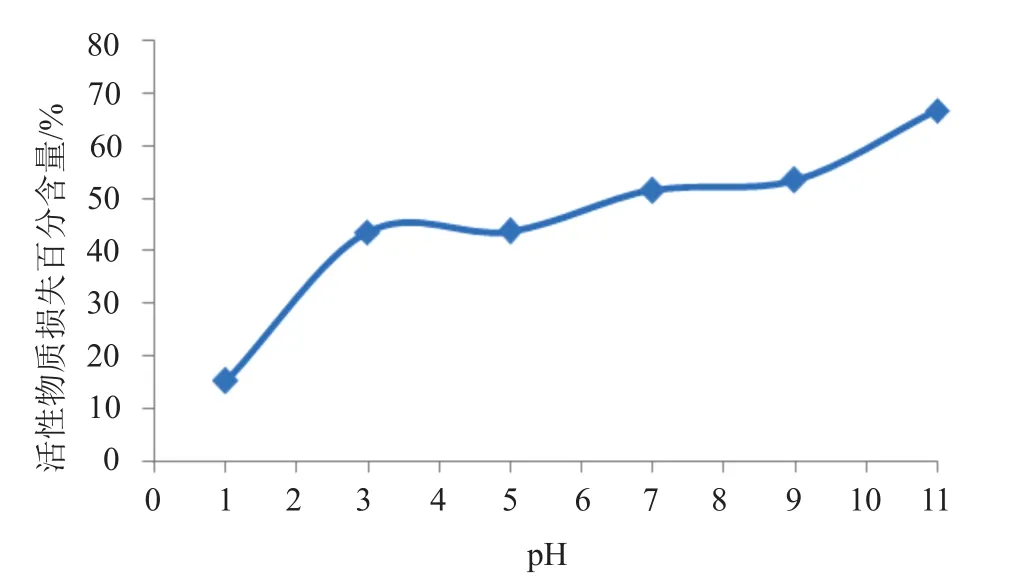
图2 在不同pH条件下DM-2树脂脱色后活性单位的损失百分含量Fig.2 Percentage of loss of active units after decolorization of DM-2 resin at different pH

图3 732树脂的静态吸附动力学曲线Fig.3 Static adsorption kinetics of 732 resin

图4 不同流速下的穿透曲线Fig.4 Penetration curves at different flow rate
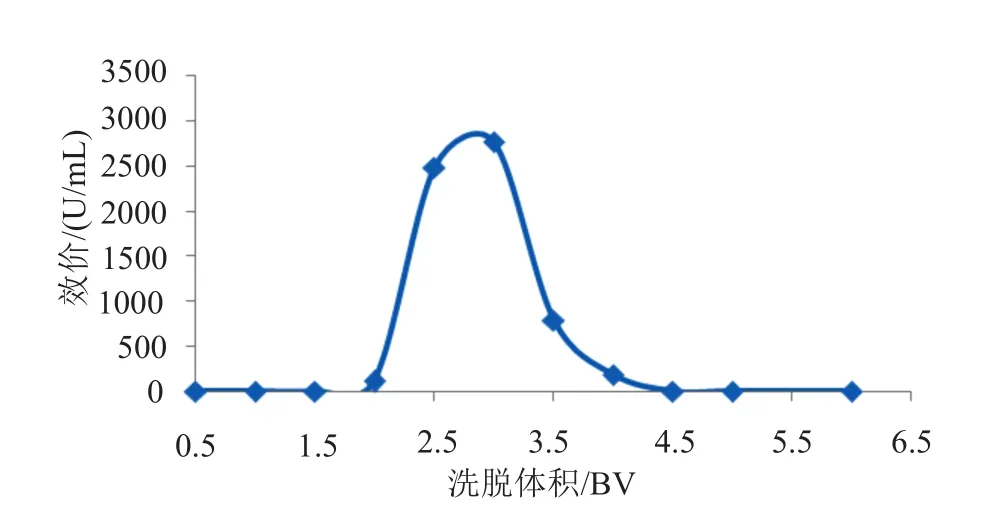
图5 732树脂的动态吸附动力学曲线Fig.5 Dynamic elution curve of 732 resin
2.5 中性氧化铝柱柱层析
将3.99g经乙醇沉淀后褐色粉末经氧化铝柱进一步精制,可以去除大部分杂质。通过生物活性跟踪可以发现活性组分主要集中在70%甲醇与50%甲醇洗脱液中。将得到的活性洗脱液进行浓缩,加入8BV甲醇后得到白色沉淀物,经干燥称重得420mg。如表1所示,产物纯度得到了进一步提升,达到86.19%。
2.6 葡聚糖凝胶柱层析
将14.6mg甲醇沉淀物进一步纯化,通过生物活性跟踪,分别将两部分收集液合并,编号A、B管,冻干后分别得到8.7和4.0mg白色粉末。
用92%H2O(0.1%TCA)+8%甲醇作流动相分别对A管、B管洗脱液进行液相检测,如图6所示,A管中物质纯度98.49%,命名为物质A;B管中物质纯度98.64%,命名为物质B。为了进一步对其结构进行鉴定,对A、B进行冻干处理,得到白色粉末。
2.7 活性物质结构解析
2.7.1 紫外可见分光光度法
波谱扫描活性物质在不同条件下检测紫外吸收光谱(图7),pH对其吸收光谱影响显著,说明其生色团中含有酸碱基团,并且其紫外特征与稻瘟菌素的紫外光谱相类似,由此推测其可能是胞嘧啶核苷类化合物[9-10]。

图6 高效液相检测化合物A和化合物BFig.6 HPLC analysis of compound A and compound B
2.7.2 化合物质谱分析
根据ESI-MS质谱(图8),由445[M+H]+和444[M+H]+峰可以推测出化合物A和B的分子量分别为444和443。
2.7.3 核磁共振波法结构鉴定
从化合物A的1H NMR(D2O)(图9)数据分析其主要化学位移,并结合分子量、氢谱、碳谱(图10)数据推测其分子式为C16H24N6O9,其C和H化学位移与文献报道的云南霉素[11]相近(表2),鉴定化合物A为云南霉素。从化合物B的1H NMR(D2O)(图11)数据分析其主要化学位移,由化合物B的13C NMR(D2O)(图12)分析,该化合物含有16个碳,结合其化学位移,分子量、氢谱、碳谱数据推测其分子式为C16H25N7O8,其C和H化学位移与文献报道的谷氏菌素[12-13]比较相近(表3)。同时由于宁南霉素与谷氏菌素的区别仅在于结构式中二肽部分丝氨酸的构型不同,所以仅通过氢谱与碳谱数据不能确定其为具体谷氏菌素、宁南霉素或两者混合物,这需要结合其他相关检测方法进一步验证。

图8 化合物A和B的质谱图Fig.8 Mass spectrum of compound A and compound B
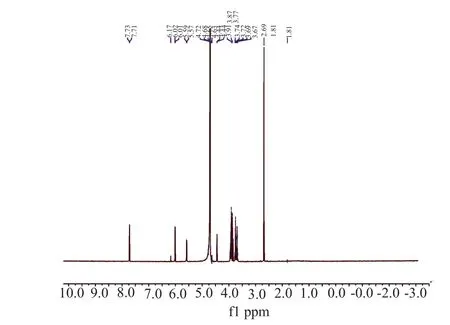
图9 化合物A的氢谱Fig.9 The hydrogen spectrum of compound A
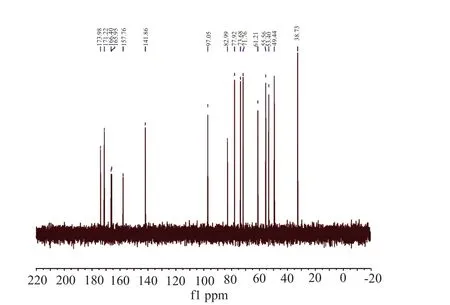
图10 化合物A的碳谱Fig.10 Carbon spectrum of compound A

表2 化合物A与云南霉素的1H和13C 化学位移数据对比Tab.2 Comparison of 1H and 13C chemical shift data for compound A and yunnanmycin

图11 化合物B的氢谱Fig.11 The hydrogen spectrum of compound B

图12 化合物B的碳谱Fig.12 Carbon spectrum of compound B

表3 化合物B与谷氏菌素的1H和13C 化学位移数据对比Tab.3 Comparison of 1H and 13C chemical shift data for compound B and gougerotin
通过对紫外光谱、质谱及核磁数据的综合分析,确定化合物A和B为核苷类化合物,其分别与文献已报道的云南霉素和谷氏菌素或宁南霉素结构一致。
3 讨论
不吸水链霉菌(Streptomyces ahygroscopicus)S91(CGMCC4.7082)能够发酵产生丰加霉素、制霉菌素、四霉素和茴香霉素。该菌种基因组中还包含谷氏菌素生物合成基因簇(GenBank: KY089035.1),但目前在该菌种的发酵产物中并未分离得到这些抗生素。说明该合成基因簇处于“沉默”状态,表达量少或者根本没有表达,从而无法检测到相应产物。
本课题使用实验室前期构建的同时阻断四霉素、制霉菌素和丰加霉素生物合成的阻断菌株不吸水链霉菌ΔttmS1ΔnysBΔtymH,发酵液中分离纯化获得云南霉素、谷氏菌素或宁南霉素。云南霉素、谷氏菌素和宁南霉素都由同一个合成基因簇编码合成,云南霉素是谷氏菌素合成的中间代谢产物,宁南霉素和谷氏菌素是立体异构体,能够相互转化。这2种或3种抗生素的产生,证明该“沉默”基因簇得到了激活。阻断其他抗生素的合成为什么会激活“沉默”基因簇,目前还不清楚,可能是由于谷氏菌素族抗生素和丰加霉素都属于核苷类抗生素,一种抗生素的基因阻断(沉默)增加了另一种抗生素前体的供应,从而提高了其产量。这种现象已有报道,如在不吸水链霉菌中阻断四霉素生物合成,同属大环多烯类抗生素的制霉菌素产量得到提高[14]。云南霉素和谷氏菌素生物合成基因簇已经被报道,因此也可以参考文献方法进一步通过基因工程的方法在不吸水链霉菌中提高相应抗生素的产量[15]。
猜你喜欢
当代水产(2022年1期)2022-04-26 14:35:30
西北农林科技大学学报(自然科学版)(2019年8期)2019-07-17 02:43:32
天然产物研究与开发(2018年4期)2018-05-07 06:47:53
中成药(2018年1期)2018-02-02 07:20:03
少儿科学周刊·少年版(2015年3期)2015-07-07 21:12:55
少儿科学周刊·少年版(2015年3期)2015-07-07 21:11:11
遗传(2015年5期)2015-02-04 03:06:55
海洋科学(2014年12期)2014-12-15 03:35:00
食品工业科技(2014年15期)2014-03-11 18:17:30
食品科学(2013年15期)2013-03-11 18:25:36
