
氮氧自由基催化醇选择性氧化反应的研究进展
2019-05-28 02:12李建微李晓梅
云南化工 2019年3期
梅 涛,李建微,李晓梅
(云南师范大学化学化工学院,云南 昆明 650500)
随着社会的发展,以绿色化学为主导思想,使合成变得更加精准和环境友好是科研工作者面临的新挑战。醇选择性氧化反应是有机合成中的一个非常重要的反应,其反应产物醛、酮等是合成药物、维生素、香料及合成纤维等复杂化合物的重要前体。因此,无论是在实验室研究,还是在工业生产中,醇选择性氧化反应都是一个非常重要的领域[1]。传统的醇氧化反应方法中,多采用化学计量的Cr(VI)、Mn(V)类无机氧化剂,这类试剂不仅成本高、有毒,且会生成大量的有害废物,显然已不符合当代绿色化工的发展要求[2-3]。
氮氧自由基具有强选择性催化氧化性能。含稳定氮氧自由基结构的化合物,在加快醛或酮转化的同时,能有效地防止过氧化成羧酸[4-5],是非常有应用前景的有机小分子催化剂。本文主要对含氮氧自由基的化合物的均相及多相催化体系催化醇选择性氧化反应的条件、优缺点及反应机理进行了综述。
1 氮氧自由基均相催化剂体系
1.1 TEMPO均相催化体系
2,2,6,6-四甲基哌啶氮氧化物 (TEMPO)被合成初始,主要用于电子自旋标定。1965年,Golubev等[6]首次报道了TEMPO可以将伯醇选择性氧化为相应的醛,活性主要源于 TEMPO结构中的NO·基团:在反应初始,TEMPO可失去一个电子被氧化为TEMPO+,在反应过程中起到氧化剂作用;反应完成后,TEMPO+又被还原为TEMPOH (图1所示)。因此,作为均相催化剂,TEMPO具有高的选择性催化氧化性能,尤其是对伯醇及小分子仲醇表现出了接近定量的产物选择性和突出的氧化催化活性[7]。
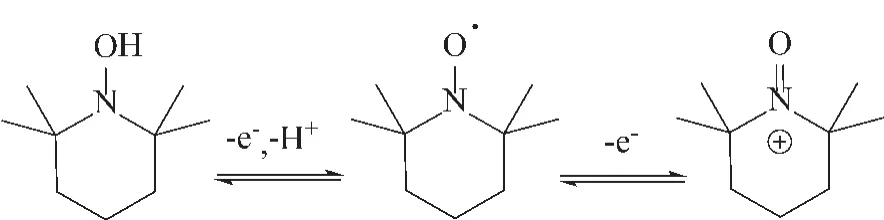
图1 不同TEMPO氧化/质子化状态Fig.1 Different oxidation/protonation states of TEMPO
1.1.1 金属参与的催化体系
金属Cu的加入可以显著提高TEMPO的催化性能。Semmelhack课题组[8]首次将金属化合物CuCl加入 TEMPO,探讨了 CuCl/TEMPO/NaClO催化体系对各类脂肪及芳香醇的催化性能影响。
研究结果显示,该催化体系更加适用于催化苄型及烯丙型伯醇类的选择性氧化,对脂肪醇催化效果不理想。随后,Ansari等[9]发现在离子液体 [Bmim]PF6中,CuCl/TEMPO体系不仅对芳香醇的氧化效果较好,对部分脂肪醇也有很好的催化活性。Ragauskas等[10]采用 Cu(ClO4)2/乙酰-TEMPO/DMAP催化剂体系,在离子液体 [Bmim]PF6中实现了各种醇的催化氧化,催化体系重复使用5次之后,活性无明显降低,但催化氧化仲醇效果不理想。以上课题组研究给出的反应机理与Golubev提出的反应机理相似。
2003年,Sheldon等[11]提出了不同的反应机理,认为整个选择性氧化反应是铜在TEMPO协助下的脱氢过程 (图 2所示),Cu(I)和 TEMPO都参与了醇的选择性氧化。研究还发现,由于仲醇甲基的存在造成很强的空间位阻,影响了TEMPO脱氢,因此 CuBr2(2,2-bipyridine)/TEMPO在碱存在下可以在室温下有氧氧化伯醇,但对仲醇没有活性[12]。 Knochel等[13,14]的研究结果显示TEMPO/Cu(I)催化体系更加适用于芳香醇和烯丙型醇类化合物的氧化,对取代的环己醇也有很好的选择性,但催化氧化仲醇的速率比伯醇慢,催化体系可重复使用 8次而保持活性不降低。
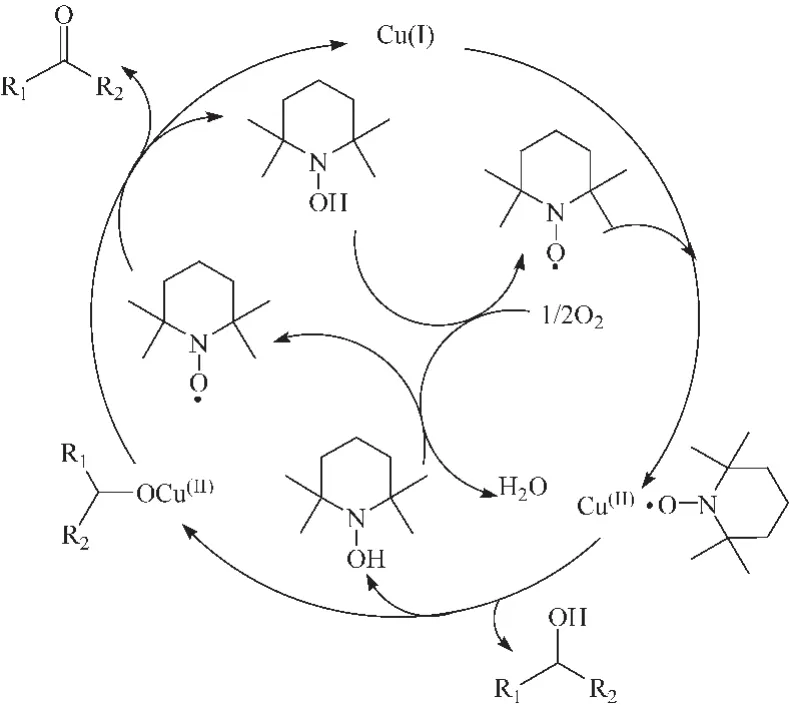
图2 CuCl/TEMPO体系催化醇氧化反应机理Fig.2 Mechanism of alcohol oxidation catalyzed by CuCl/TEMPO
Stahl课题组[15]在前人研究的基础上在催化体系中引入 (bpy)CuI。 (bpy)CuI/TEMPO/NMI催化体系可以在空气或O2中常温催化氧化除苄型、烯丙型以外的丙炔型醇,拓宽了氮氧自由基催化不同类型醇底物的范围。该课该题组认为反应过程包括两步:第一步,被活化后的 Cu2O2与TEMPO结合成为CuII-OH中间产物,该中间产物不稳定,随即转化为CuII-醇盐,CuII-醇盐才是真正意义上的催化剂 (图3所示)。随后,该课题组[16]又考察了三氟甲磺酸亚铜 (CuOTf)/2,2-联吡啶 (-bpy)/TEMPO/N-甲基咪唑 (NMI)组成的催化体系对不同醇选择性氧化性能的影响,得到了最佳的反应条件,即:以空气或O2为氧源 , 5%bpy、 5%CuIOTf、 5%TEMPO、 10%NMI(均为摩尔分数)条件下,催化体系可以催化氧化除苄型、烯丙型以外的丙炔型以及脂肪伯醇为相应的醛,但是,脂肪族伯醇若要全部转化,则需要加热至50℃。
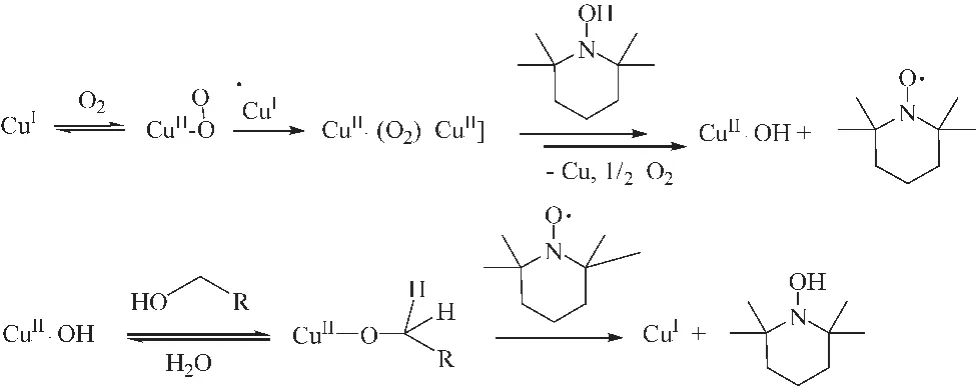
图3 (bpy)CuI/TEMPO/NMI催化醇氧化反应机理Fig.3 Mechanism of alcohol oxidation catalyzed by (bpy)CuI/TEMPO/NMI
Liaigre等[17]研究了固载有席夫碱铜配合物的微球 [Cu(BABAP)2]-CPGMA与TEMPO构成共催化体TEMPO/[Cu(BABAP)2]-CPGMA催化体系的催化性能,苯甲醇的转化率高达93%。其催化机理为见图 4所示。随后,Figiel等[18]报道了在碱性水溶液中,将TEMPO与2-吡咯甲醛碳二亚胺和铜复合物组成新的催化体系催化氧化醇,结果发现苯甲醇可以被定量的转化为苯甲醛,产物的选择性也很高。然而,这种配体合成困难,原料昂贵,反应体系也要加入额外的碱。
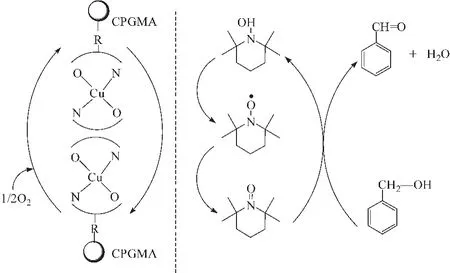
图4 共催化剂TEMPO/[Cu(BAAP)2]-CPGMA催化氧化苯甲醇的机理Fig.4 Mechanism of Catalytic Oxidation of Benzyl Alcohol by Cocatalyst TEMPO/[Cu(BAAP)2]-CPGMA
综上,Cu(I)的加入显著提高了TEMPO的催化性能,拓宽了体系催化不同类型醇底物的范围。除了Cu(I),其它金属也实现了优异的催化氧化性能。
研究发现[19],贵金属Ru与TEMPO组成的催化体系,活性更高。1%RuCl2(PPh3)3和 3%TEMPO组成的共催化体系在100℃、10 atm、氯苯为溶剂的条件下,能有效的氧化各种脂肪伯醇、仲醇、苄醇和烯丙醇为相应的醛/酮化合物,产物选择性>99%,尤其对仲醇的催化性能更高。反应过程中RuCl2(PPh3)3与醇反应生成相应的羰基化合物 (RuH2L3),TEMPO的作用是促使Ru催化剂再生,而TEMPO被还原为 TEMPOH(图5所示)。另外,该体系对含有N、S、O等杂原子官能团的醇催化活性欠佳,可能是由于这些原子对Ru有一定的配位作用。但是Ru是贵金属,不满足绿色化学经济要求,而且存在重金属释放到环境中的问题,不易工业应用。
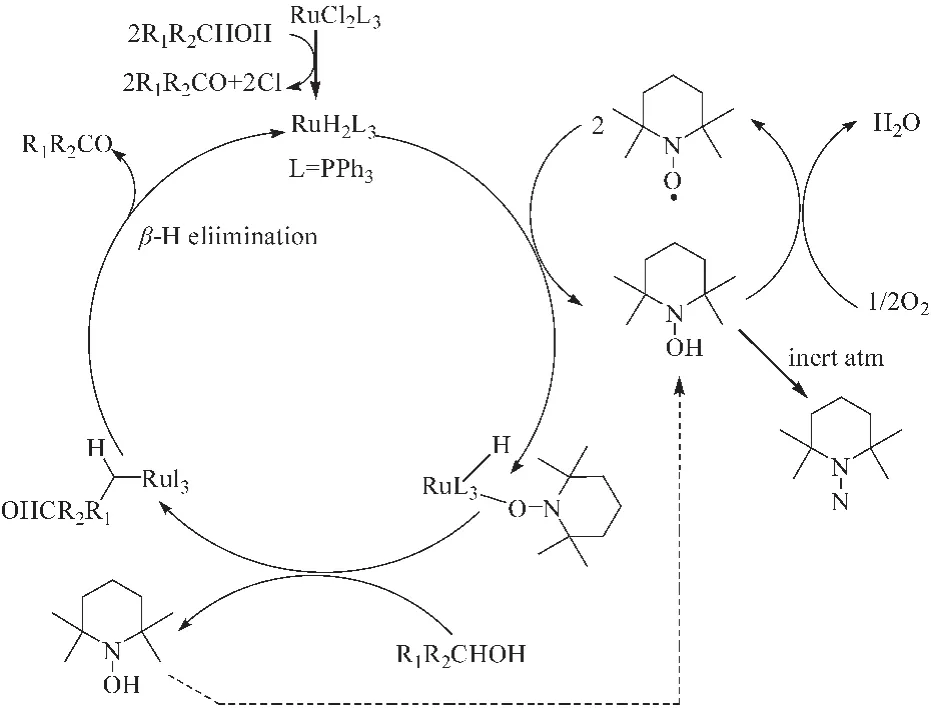
图5 RuCl2(PPh3)3/TEMPO催化醇的氧化反应机理Fig.5 Mechanism for aerobic oxidation of alcohols catalyzed RuCl2(PPh3)3/TEMPO
1.1.2 无过渡金属参与的催化体系
无金属参与的醇选择性氧化反应中,由于没有金属参与,不存在重金属残留的问题,也不存在重金属释放到环境中的问题,在醇氧化反应中有很大的发展空间。
Neuwann等[21]发现杂多酸 H5PV2Mo10O40/TEMPO催化体系与贵金属 RuCl2(PPh3)3/TEMPO催化性能相当,但更加经济。Anelli[20]课题组研究发现1%TEMPO、10%NaBr(均为摩尔分数)组成的 TEMPO/NaBr催化体系在 pH为 9的CH2Cl2溶剂中,氧化剂NaClO作用下,可以将伯醇和仲醇选择性的快速氧化为相应的醛或酮。课题组认为的反应机理与Golubev课题组相似,是由次氯酸盐和NaClO激发的TEMPO+/TEMPOH之间两个电子氧化还原循环过程,TEMPO+才是实际上醇氧化过程的氧化剂 (图6所示)。需要注意的是该催化体系中的NaBr可与次氯酸盐反应生成次溴酸盐,与次氯酸盐相比,次溴酸盐更易于与TEMPOH反应。
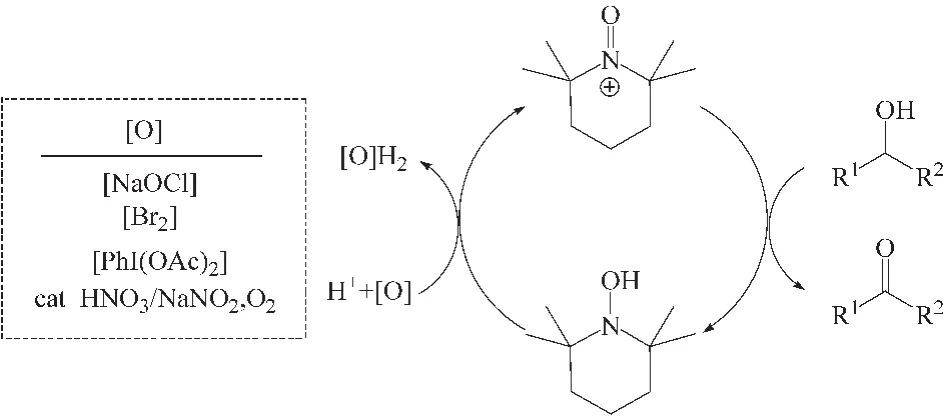
图6 TEMPO/NaClO/NaBr催化氧化机理Fig.6 Mechanism of alcohol oxidation catalyzed by TEMPO/NaClO/NaBr
2004年,Liang等[22]研究了TEMPO/NaNO2/Br2催化体系的催化氧化性能,该体系利用NaNO2产生的 NO活化分子氧,可以将多种芳香醇定量、高选择性的氧化为相应的醛或酮,底物中的N、S杂原子不影响反应的进行。但是该体系对脂肪伯醇只具有中等程度的选择性,氧化产物中只有酸或酯的产生。这是第一个实现无过渡金属参与的以分子氧为氧化剂的TEMPO催化醇氧化反应体系,开创了TEMPO催化醇氧化的新领域。
2005年,Liu等[23]报道了O2为末端氧化剂的TEMPO/NaNO2/Br2催化体系,反应机理如图 7所示,该体系以二氯甲烷为溶剂,在80℃、0.4~0.9 MPa空气的反应条件下,能高选择性氧化伯仲醇及含杂原子的醇为相应的醛或酮。He等[24]研究发现,以1,2-二氯乙烷为溶剂,该催化体系在0.2 MPa O2和80℃的条件下,高效氧化苄基醇和脂肪伯仲醇为相应的醛或酮。
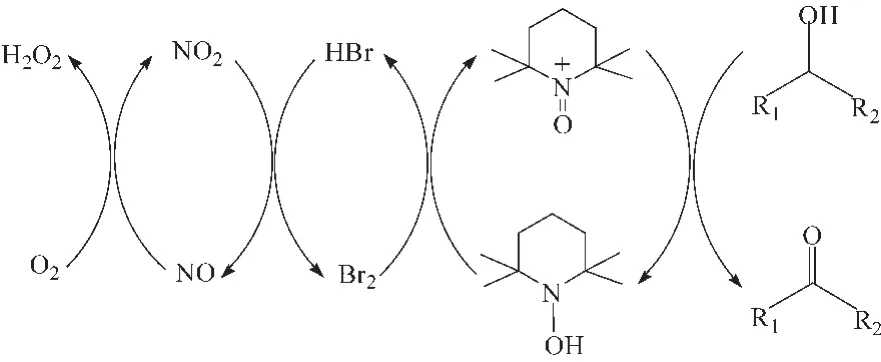
图7 TEMPO/NaNO2/Br2催化醇的有氧氧化反应机理Fig.7 Mechanism for aerobic oxidation of alcohols catalyzed byTEMPO/NaNO2/Br2
综上,金属、杂多酸等物质与TEMPO组成的均相催化体系,在金属与TEMPO的协同作用下,展现出了高的醇选择性催化氧化性能。除了TEMPO,其它具有 NO·结构的化合物也同样显示了高的醇催化氧化性能。
1.2 TEMPO衍生物催化剂
2008年,Ragauskas课题组[25]设计合成了四种结构的TEMPO衍生物,其中催化活性最好的是 4-苄胺基-TEMPO,在温和条件下,其与CuBr2、含氮配体组成的催化体系可以将一系列的苄醇高效氧化成相应的醛或酮。最佳反应条件为:以3.0 mmol苯甲醇为模板底物,3%(摩尔分数) 的4-苄胺基-TEMPO用量,溴化铜2%(摩尔分数),80℃,6 mL H2O的条件下反应 5 h,苯甲醇的转化率达到98%,苯甲醛的选择性也达到99%以上,对底物的普适性也十分理想,该催化体系是一个不需要另外加入任何碱、含氮配体、有机溶剂的简单高效催化体系。
Shinichiro等[26]制备了TEMPO多种衍生物,结果发现:4-甲氧基-TEMPO和乙酰氨基-TEMPO表现出了较高的氧化效率,4-羟基-TEMPO的氧化效率很低。4-乙酰氨基-TEMPO/NaClO/NaClO2氧化机理如图 8所示[27],与TEMPO/NaClO/NaBr催化体系相比,4-乙酰氨基-TEMPO/NaClO/NaClO2的可控性更好[28]。STAHL课题组[29]成功合成了AcNH-TEMPO催化剂,研究了催化剂体系对各种醇的催化氧化性能,得到适宜的催化剂用量为5%,以O2为氧源,在10%HNO3和10%HCl(均为摩尔分数) 的共同作用下,产物酮的收率高达96%。
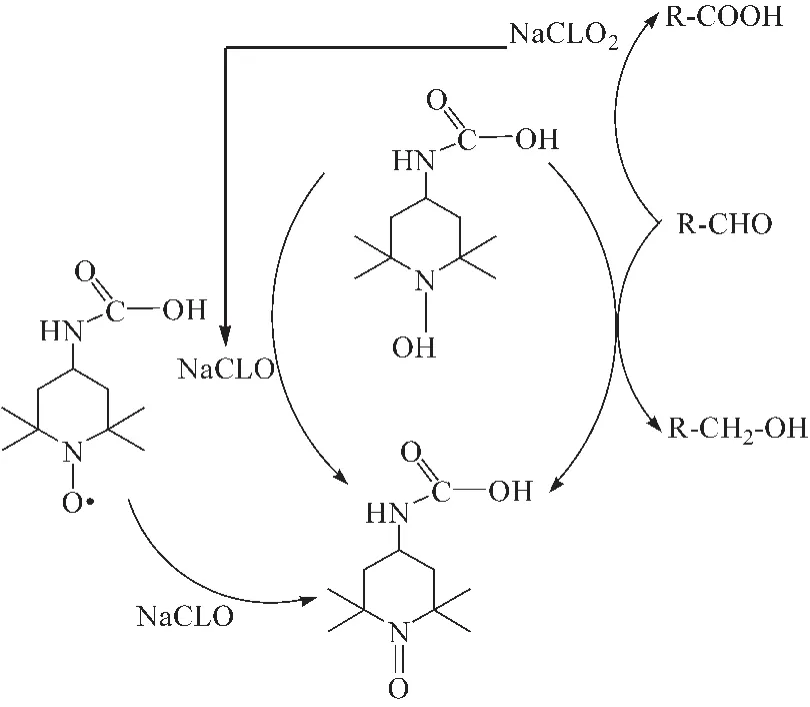
图8 4-乙酰胺基-TEMPO/NaClO/NaClO2催化体系的氧化机理Fig.8 Oxidation mechanism of 4-acetamido-TEMPO/NaCLO/NaCLO2system
由TEMPO衍生出来的其它含氮氧自由基结构的化合物也同样显示了优异的醇氧化催化性能。Yoshiharu课题组[30]以TEMPO为原料,设计、合成出的AZADO和1-Me-AZADO两种含氮氧自由基的双环结构化合物 (图9所示),随后又制备了ABNO、ketoABNO和 F-AZADO几种类似的双环结构化合物[31]。结果显示,以上双环结构的化合物比TEMPO具有更高的醇氧化催化性能,而且在一定程度上拓宽了催化醇的类型。
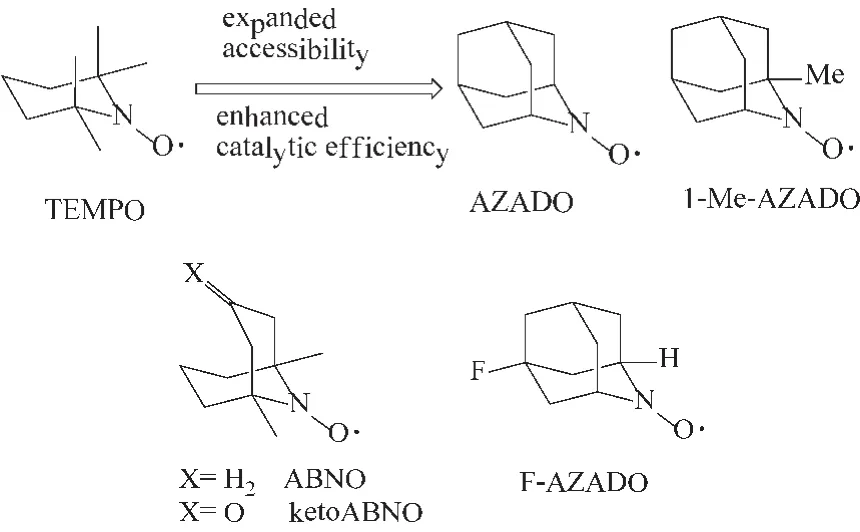
图9 AZADO、1-Me-AZADO、ABNO、ketoABNO和 FAZADO结构示意图Fig.9 The structure of AZADO,1-Me-AZADO,ABNO,ketoABNO and F-AZADO
Stahl课题组[32]在对 CuOTf/bpy/TEMPO/NMI体系研究的基础上,继续考察了ABNO、ketoABNO和AZADO几种双环结构化合物与CuOTf/bpy/NMI组成的催化体系对不同类型醇催化速率的影响规律。结果显示 CuOTf/TEMPO对不同类型醇的催化速率明显不同,反应速率从高到低依次为:苄型伯醇 >脂肪族伯醇/苄型仲醇≫脂肪族仲醇/大空间位阻苄型仲醇;相比之下,CuOTf/ABNO体系对以上几种醇的催化速率几乎相等,且催化速率远远高于CuOTf/TEMPO。课题组认为,主要原因可归结为双环结构低的空间位阻,氧化过程的完成需要CuOTf和氮氧自由基的共同作用[33]。
Iwabuchi课题组[34]研究了 (bpy)Cu/AZADO的氧化性能。结果显示,加入Cu(I)后的催化效果明显优于 Cu(II), (bpy)Cu/AZADO高化学选择性的机理可归结于:Cu(I)作为催化剂时,可与O2作用生成中间产物LnCuII-OH碱,该碱可与醇结合生成CuII-OH醇盐,醇盐与TEMPO之间发生H原子的转移,最后生成相应的羰基化合物 (反应机理如图10所示),金属和氮氧自由基两者协同作用能达到理想效果。
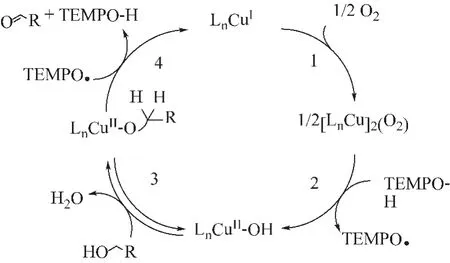
图10 (bpy)Cu/TEMPO催化醇氧化反应机理Fig.10 Mechanism of alcohol oxidation catalyzed by (bpy)Cu/TEMPO
综上,含NO·的均相催化体系显示出了优异的醇选择性氧化催化性能。但是以上方法中多使用有毒溶剂、反应条件不环境友好,另外,NO·体系为均相催化体系,存在后期难与产物分离、不易于重复使用等问题,限制了其工业应用。因此,为克服上述困难,人们尝试将均相化催化剂负载到各种载体上,使其易于分离。常用的载体有碳纳米管[35]、羟基磷灰石[36]、三氧化二铝[37]、沸石等[38],其中用于固载氮氧自由基的载体主要有不溶性有机高分子、介孔材料和金属有机骨架等。
2 氮氧自由基多相催化剂
2.1 多孔材料为载体
多孔材料表面的Si-OH可以和活性中心以共价键牢固结合,达到固定活性中心的目的。Kimoto等[39]将TEMPO固载到多孔硅珠 (PSB,porous silica beads)无机材料表面上,再将NOX气体吸附到多孔硅珠的孔道内得到PSB-TEMPONOx催化剂。活性测试结果显示,室温下反应4 h后,苯甲醇转化率达到 100%,选择性达到99%以上。反应完成之后,经过过滤和旋蒸操作,可以使产物的纯度达到99%,催化剂体系可以重复使用多次。整个氧化反应发生在催化剂的孔道内部,包括如图11所示的三个阶段:1)醇底物分子进入孔道内部;2)底物被氧化为羰基化合物;3)羰基化合物扩散出孔道进入溶剂中。

图11 PSB-TEMPO-NOX多相催化体系下反应机理Fig.11 The proposed mechanism for PSB-TEMPO-NOX catalytic system
Cirminna等[40]将TEMPO固载到粒度为 60 μm、孔径为 10 nm的硅胶上,制备了非均相催化剂SA-TEMPO (图 12所示),吸附在硅胶孔道中的NOx气体为共催化剂。空气为氧化剂,SATEMPO/NOx催化体系可以在2.5 h内将苯甲醇完全转化为苯甲醛。该催化系统的优点是二氧化硅具有足够的机械强度,在磁力搅拌下不会开裂,NOx仍然保留在二氧化硅孔道中,达到催化剂的重复使用。需要注意的是该催化体系在催化氧化芳香类仲醇时的反应时间较长,催化氧化脂肪类伯醇效果没有芳香类醇好,而且在氧化对甲氧基苯甲醇时会有多种副产物产生。
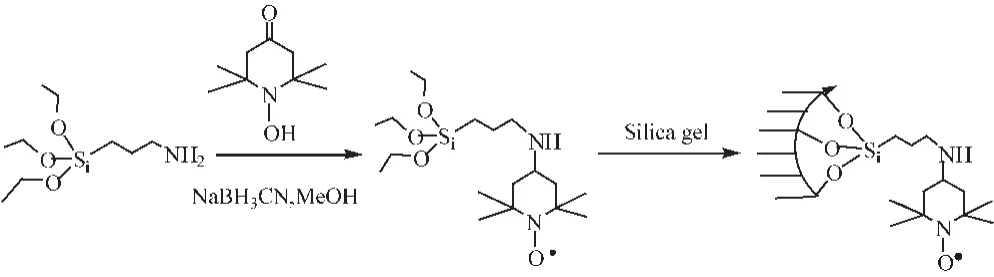
图12 硅胶-TEMPO的制备Fig.12 Preparation of silica gel-TEMPO
介孔材料(孔径介于 2~50 nm) 具有均一且可调的孔径、高度有序且稳定的骨架结构、具有一定壁厚且易于掺杂的无定型骨架以及比表面积大且可修饰的内表面等特征,其大尺度的孔道极方便于传质过程的进行,故在多相催化领域,介孔材料(尤其是SBA-15和MCM-41) 作为催化剂载体[41]为大分子的大宗化学品和精细化学品制备与处理提供了更加经济和环境友好的技术途径。
2001年,Brunel等[42]报道了介孔分子筛MCM-14作为载体,分别与 4-hydroxy-TEMPO和4-carboxy-TEMPO反应得到两种固载型的TEMPO多相催化剂 (图 13所示),采用该催化剂与CuCl组成催化体系催化苯甲醇的选择性氧化,但催化活性大不如负载前,反应过程中没有TEMPO的流失。
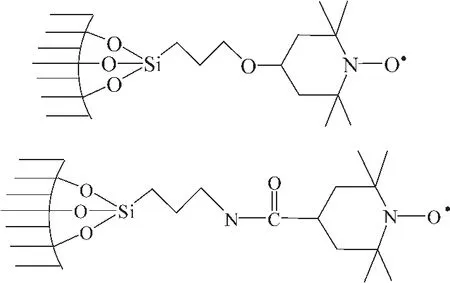
图13 TEMPO-ether-MCM-41和TEMPO-amide-MCM-41的结构Fig.13 The structures of TEMPO-ether-MCM-41 and TEMPO-amide-MCM-41
Babak课题组[43]将TEMPO固载于介孔材料SBA-15,用于催化苯甲醇及其衍生物的氧化。固载后的催化剂具有优异的重复使用性。Zhang等人[44]也成功将 TEMPO固载于 SBA-15,以O2为氧化剂、常压、FeCl3的共同作用下,苯甲醇的转化率为94.5%,但是反应中仍需加入有毒溶剂甲苯及NaNO2。但是这类后固载型催化剂存在活性中心分布不均匀、反应过程中催化中心易流失、催化剂不能重复使用等问题。
2.2 聚合物为载体
Sheldon等[45]以低聚物稳定剂 Chimassorb 944为载体,将TEMPO固载制成多相催化剂PIPO(图 14所示),制备的多相催化剂活性比 TEMPO更高,而且该催化剂在非极性溶剂中不易溶,利用这一特性可将催化剂回收利用。
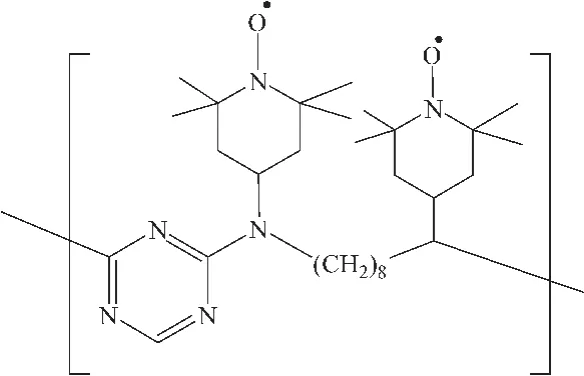
图14 PIPO结构示意图Fig.14 The structure of PIPO
2005年,Maurizio等[46]将 TEMPO固载到聚乙二醇表面,得到非均相催化剂 PEGTEMPO。PEG-TEMPO与 Co(NO3)2或 Mn(NO3)2组成的共催化剂体系对伯醇具有良好的催化活性,对仲醇也有一定的催化活性,但转化率较低。该方法开拓了实现 TEMPO固载化的新思路。见图15。
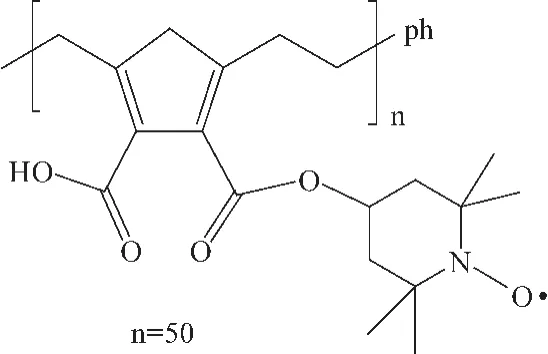
图15 烯烃聚合反应得到的固载TEMPO催化剂Fig.15 Olefin polymerization to obtain a supported TEMPO catalyst
Parlett[47]以交联聚苯乙烯 (CPS)微球为载体,制备了非均相固体催化剂 TEMPO/CPS。以乙酸为溶剂,固载后的TEMPO/CPS与助催化剂Fe(NO3)3的物质的量比为 1∶1.2的条件下,主催化剂含量为11%时,该组合体系能在温和条件下将1-苯乙醇催化氧化为苯乙酮,转化率为96%,且微球的循环使用性能良好,相应的催化机理如图16所示。
2.3 金属有机骨架为载体
Susumu课题组[48]将含有氮氧自由基活性中心的功能性有机配体 DPIO (图 17,左)进行组装,首次合成了新型的氮氧自由基修饰的金属有机骨架 FRPCP(图 17,右)。FRPCP与 TBN组成的催化体系,可将苯乙醇氧化成苯乙醛,苯乙醛产率为94%,在整个反应过程中氮氧自由基无流失。但是该反应需要在80℃条件下进行、反应时间长,而且利用合成金属骨架实现氮氧自由基固定的合成技术步骤繁琐,FRPCP产率极低。
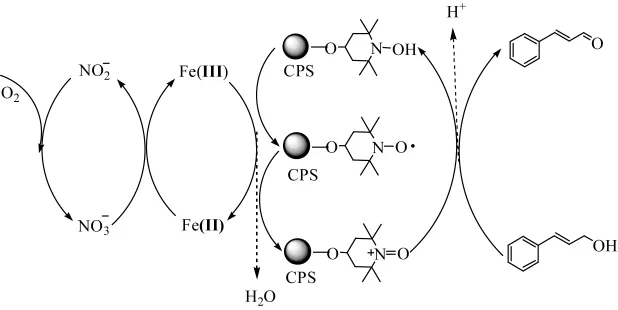
图16 TEMPO/CPS与Fe(NO3)3的组合催化剂催化氧化机理Fig.16 Reaction Mechanism of EMPO/CPS and Fe(NO3)3Catalysts for Catalytic Oxidation
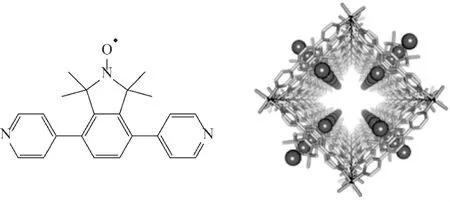
图17 DPIO有机配体 (左)与FRPCP(右)多孔配位聚合物Fig.17 DPIOorganic ligands and FRPCP porous coordination polymers
综上所述,对于均相NO·催化体系催化催化醇氧化反应研究较多,但是对于多相NO·催化体系的研究只是在初级阶段,目前尚未找到性能优异的材料及方法进行NO·活性中心有效固载。NO·多相催化剂研究中普遍存在合成方法较复杂、固载后的催化剂存在活性不高或活性中心易流失等问题。因此,在发挥氮氧自由基高催化活性、选择性的同时,寻找新的氮氧自由基固化材料及固载方法尤为重要。在绿色化学指导下,将催化氧化活性高的NO·固载制备高效的多相催化剂,是发挥氮氧自由基催化性能的有效措施。
3 结语
本文针对不同类型的均相及多相的含NO·催化剂体系催化醇选择性氧化反应进行了总结,对该反应机理的研究结果进行整理。NO·作为小分子催化活性中心具有广泛的应用前景,而醛、酮等是合成药物、维生素、香料及合成纤维等复杂化合物的重要前体。均相催化剂的效能较高,但存在后期分离问题;多相催化剂固载材料的发展方向逐渐由简单挑选不同的载体、助剂等方式改变为创新性的材料和制备方法,如聚合物、FRPCP等载体。固载后的催化剂尚存在合成步骤繁琐,活性不是很高等问题。因此,该研究还有很广大的发展空间,希望通过新型催化剂的合成开拓更广的醇氧化反应催化剂并为其工业化做指导。
猜你喜欢
农业资源与环境学报(2022年3期)2022-05-26
建材发展导向(2021年14期)2021-08-23
农业资源与环境学报(2021年4期)2021-07-30
太原科技大学学报(2020年3期)2020-06-22
中国煤层气(2019年2期)2019-08-27
第一财经(2019年8期)2019-08-26
中南民族大学学报(自然科学版)(2019年1期)2019-04-04
安徽医科大学学报(2015年9期)2015-12-16
支点(2015年11期)2015-11-16
学习月刊(2015年14期)2015-07-09
