
高压下BeP2晶体结构与物性的理论研究
2019-05-25 00:48高文泉李鑫马雪姣刘艳辉
延边大学学报(自然科学版) 2019年1期
高文泉, 李鑫, 马雪姣, 刘艳辉
( 延边大学 理学院, 吉林 延吉 133002 )
近年来,二元化合物在高压下因表现出不同寻常的物理性质,引起了研究者的注意.在二元化合物中,碱土金属磷化物通常有着较大的激子半径和窄带隙,使其在光电子、探测器、红外传感器、太阳能电池、超声波倍增器和霍尔发生器中得到应用[1-6].铍元素由于质量轻、弹性模量高和热稳定性好,目前已成为重要的战略金属材料[7].但是,由于铍元素的化学性质较为活泼,且铍化物毒性较强,这给实验上的研究带来了很大的难度.
1935年,Stackelberg等首次研究了铍磷体系,经过测定一种方铁锰矿结构的晶格参数确定了该物质的结构为Be3P2[8].1975年,El-Maslout等对该体系进行研究时发现其有两种化合物存在,即Be3P2和BeP2[9-10].1976年,L’Haridon等[11]制备了BeP2单晶,并使用单晶衍射方法研究了其晶体结构,其晶格参数为a=3.546 Å,b=3.546 Å,c=15.010 Å,配位数为4, 空间群为I41/amd.2006年,Cheng等对p-GaP薄膜进行快速热退火(500 ℃)时发现了一种新型的磷化铍(BeP)离子化合物[12].2018年,Li等预测了二维BeP2单层,结果表明其具有压缩诱导的狄拉克半金属材料性质[13].目前为止,学者们通过实验已经得到了BeP2稳定的常压相结构,但对其高压下的结构还未见相关文献的报道;基于此,本文采用第一性原理的计算方法,结合CALYPSO软件,对高压下的BeP2体系结构进行理论预测和研究.
1 计算方法
使用基于粒子群优化算法的CALYPSO软件包[14],对0~100 GPa压强范围内的BeP2晶体结构进行随机搜索.搜索模拟所采用的晶胞数为2倍胞和4倍胞.结构优化采用基于密度泛函理论的VASP软件包[15],电子间的交换关联势能函数采用广义梯度近似Generalized Gradient Approximation(GGA)下的Perdew-Burke-Ernzerh(PBE)交换关联泛函,赝势采用全电子投影缀加平面波赝势[16],Be原子的价电子是2s2, P原子的价电子是3s23p3.为了确保体系能量收敛的精度小于1 meV/atom,需要对体系能量的收敛进行测试,测得平面波的截断能为600 eV.布里渊区的k点网格采用Monkhorst-Pack取点方式,网格间距为0.2 nm-1,选取10-5eV作为自洽能量收敛最小值,将优化应力收敛值设置为0.001 eV/Å.采用直接超晶胞的方法,应用PHONOPY软件,计算声子色散关系.
2 结果与讨论
2.1 晶体结构的预测
对预测得到的晶体结构进行优化,计算时温度取0 ℃.G=H-TS,因此可以用体系焓值的变化来代替自由能.绘制体系的焓差值随压强变化的曲线,得BeP2晶体结构的热力学稳定区间,如图1所示.从图1可以得出:在常压时,空间群为I41/amd的四方结构,焓值最低,记为α-BeP2相.当压强增大到30.1 GPa时,BeP2的晶体结构发生第1次相变,由空间群为I41/amd的α-BeP2四方结构相变为空间群为P43212的四方结构相,记为β-BeP2相,其稳定存在的压力区间为30.1~35.4 GPa.当压强增大到35.4 GPa时,BeP2的晶体结构发生第2次相变,由空间群为P43212的四方结构相变为空间群为Imma的正交结构相,且此结构稳定存在到100 GPa,将该结构记为γ-BeP2相.图1插图(a)中绘制了α-BeP2相、β-BeP2相和γ-BeP2相的体积随压强的变化曲线.观察曲线发现,在3相存在的稳定热力学区间内,晶体的体积随压强的增大而减小,其变化呈线性变化趋势.在BeP2相变的过程中,在30.1 GPa和35.4 GPa时伴有体积的塌缩,其塌缩率分别为7.1%和10.9%,这种结构相变属于一级相变.

图1 BeP2的热力学焓差图(插图为体积随压强的变化曲线图)
预测得到的α-BeP2相、β-BeP2相和γ-BeP2相的晶体结构如图2所示,优化后的BeP2平衡态晶格常数和原子位置如表1所示.图2(a)为在压强为0 GPa时预测得出的α-BeP2四方相的晶体结构,其空间群为I41/amd.该结构内部每个Be原子被4个P原子包围,组成四面体,且每个P原子有2个Be和2个P相邻.Be—P的键长为2.137 Å, P—P的键长为2.283 Å.晶格常数为a=b=3.582 Å,c=14.994 Å,α=β=γ=90.0°, 其中Be原子的Wyckoff占位为4a(0.500,-0.500,0.500),P原子的Wyckoff占位为8e(1.000,0.000,0.672).本文预测得到的常压相结构与文献[11]得到的BeP2结构一致,这表明本文所采用的晶体结构预测方法以及计算参数的选择是准确可靠的.图2(b)所示为在压强30.1 GPa下预测得到的β-BeP2四方相的晶体结构,其空间群为P43212.该结构中每个Be原子和最近邻的4个P原子组成四面体.晶格参数为a=b=4.997 Å,c=5.748 Å,α=β=γ=90.0°, 其中Be原子的Wyckoff占位为4a(0.074,0.074,0.000),P原子的Wyckoff占位为8b(0.647,0.831,0.249).图2(c)所示为在压强35.4 GPa下预测得到的γ-BeP2正交相的晶体结构,其空间群为Imma.该结构中每个Be原子和最近邻的6个P原子组成八面体结构.晶格参数为a=4.301 Å,b=3.937 Å,c=7.348 Å,α=β=γ=90.0°, 其中Be原子的Wyckoff占位为4e(0.000,0.250,0.927), P1原子的Wyckoff占位为4e(0.500,0.750,0.718), P2原子的Wyckoff占位为4e(0.500,0.250,0.895).
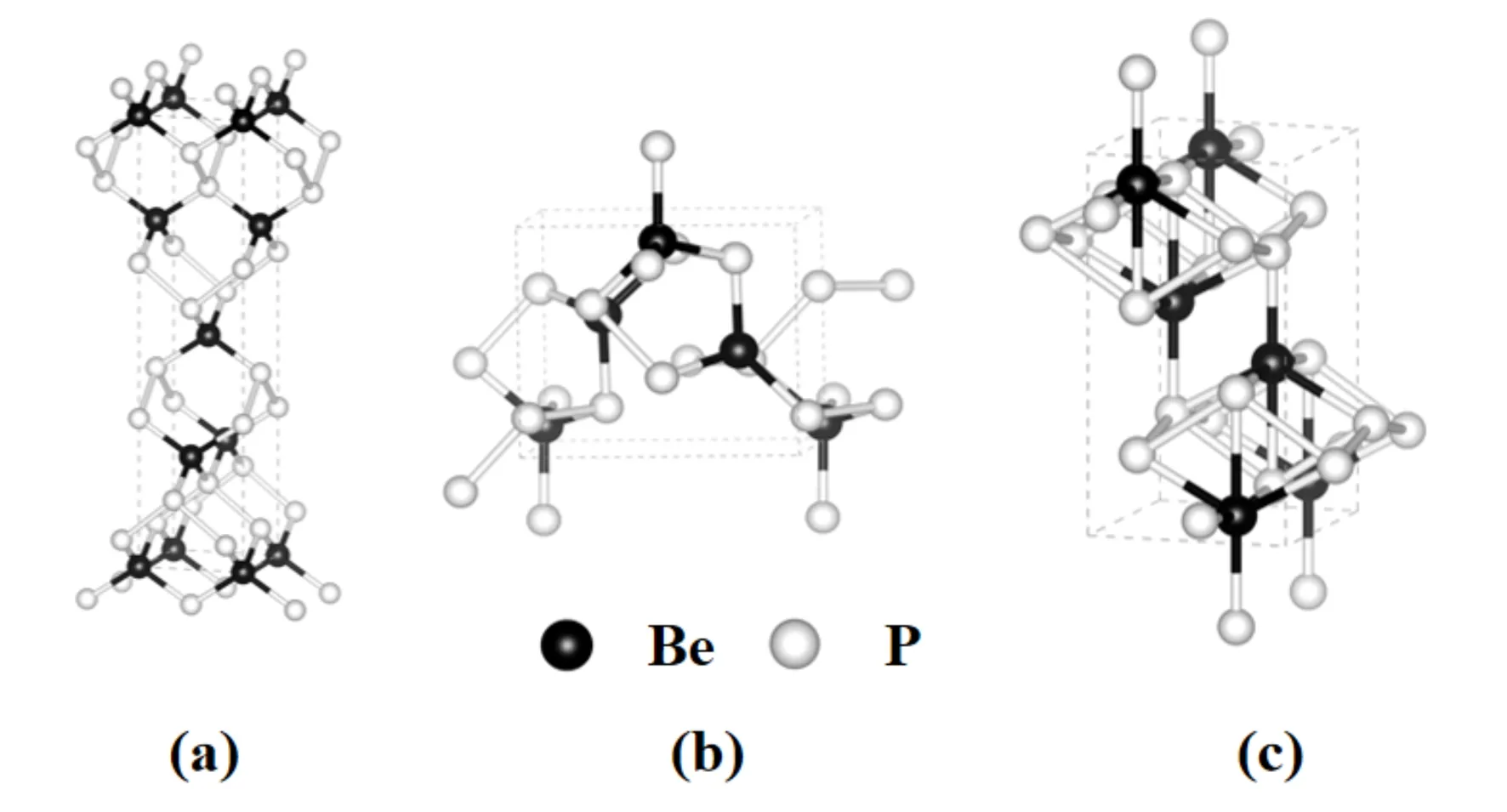
图2 α -BeP2相、β -BeP2相和γ -BeP2相的晶体结构
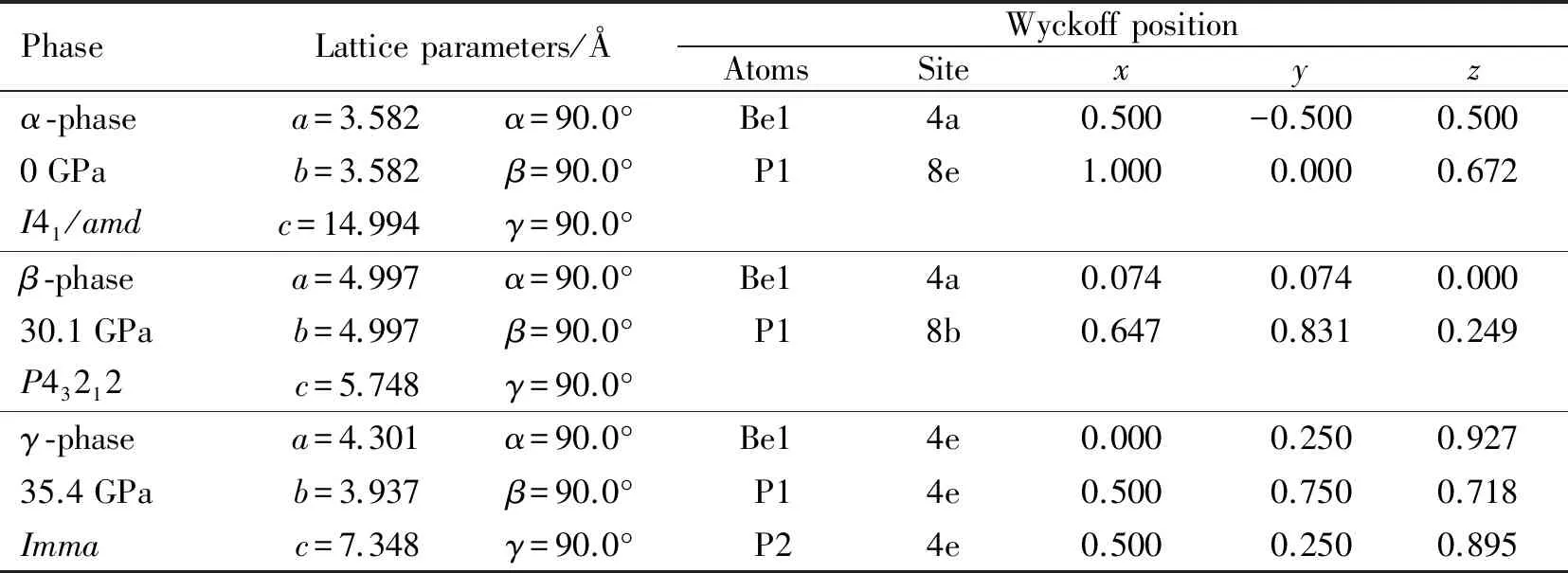
表1 α -BeP2相、β -BeP2相和γ -BeP2相的晶格参数和原子位置
2.2 晶体结构的热力学和动力学稳定性
材料结构的稳定性是判定高压结构的重要理论依据.本文通过形成焓的计算公式(1)计算α-BeP2相、β-BeP2相和γ-BeP2相(3相)结构在压强0 GPa、30.1 GPa和35.4 GPa下的形成焓.
ΔH=(HBeP2-HBe-2HP)/3.
(1)
公式(1)中,HBeP2表示3相结构在稳定压强区间内的形成焓,HBe和HP表示相应压强下的Be原子和P原子的焓值.若计算得出的焓值ΔH<0,则说明结构是热力学稳定的.经计算,得预测结构的焓值分别为-0.425、-0.239 eV/atom和-0.145 eV/atom,这说明α-BeP2相、β-BeP2相和γ-BeP2相具有热力学稳定性.
为了进一步确定BeP2晶体结构的动力学稳定性,分别计算3相结构的声子色散关系.晶格结构稳定的条件是简正声子频率都应是有限的实值[17],若为虚值,则表明材料在布里渊区发生了声子软化现象,晶格结构不稳定.3相在相应稳定热力学区间内的声子色散关系和声子态密度(PHDOS)如图3所示.绘制声子谱并进行分析表明,3相结构在整个布里渊区高对称点处的所有简正声子频率都是正值,没有出现虚频现象,由此表明BeP2高压相结构具有动力学稳定性.通过分析3相结构的声子态密度发现,在高频区域主要是由Be原子贡献,在低频区域主要是由P原子贡献.
2.3 晶体结构的电子性质
为了更好地分析BeP2基态结构的电子性质,计算BeP2各相的能带结构,结果如图4所示(图中虚线代表通过密度泛函理论画出的能带,实线代表通过HSE杂化泛函修正后的能带).由于采用密度泛函理论的方法计算带隙会有误差[18](通常会低估结构的带隙),因此本文采用HSE杂化泛函[19]的方法对PBE-GGA进行计算.
常压下,α相修正后的能带图如图4(a)所示.α-BeP2相的带隙在布里渊区G高对称方向发生明显打开,显现出半导体特性.经计算,导带底和价带顶间有0.457 eV的直接带隙,这表明α-BeP2是一种窄带隙半导体.在压强为30.1 GPa下,β-BeP2相的能带结构如图4(b)所示.β-BeP2相的带隙打开程度变大,带隙为0.975 eV,表明β-BeP2相也是一种窄带隙半导体.在压强为35.4 GPa下,γ-BeP2相的能带结构如图4(c)所示.γ-BeP2相的导带与价带依然相互相叠,在费米能级处出现了电子填充,由此表明γ-BeP2相具有金属性.总体看,α-BeP2相与β-BeP2相为半导体,γ-BeP2相为金属,即BeP2体系发生了从半导体到金属性质的转变.

图3 α -BeP2相、β -BeP2相和γ -BeP2相的声子色散关系与投影态密度

图4 α -BeP2相(0 GPa下)、β -BeP2相(30.1 GPa下)和γ -BeP2相(35.4 GPa下)的PBE能带图和HSE修正后的能带图
为了确定BeP2晶体的成键类型,计算BeP2晶体结构的电子局域函数(ELF),等值面选取的数值为0.87.图5分别为3相的电子局域函数图.观察图5可知,在Be原子和P原子之间有着明显的电子局域现象,电子云集中分布在P原子周围,并偏向于P原子,且P原子和P原子周围也存在电子,这表明Be原子与P原子之间、P原子与P原子之间存在着共价键.为了更清晰地描述原子间的电子特点,计算BeP2的Bader电荷转移[20]和各相中P原子到Be原子转移的电荷,结果见表2.从表2可知,Be原子是受主,P原子是施主,3相的结构均发生了P原子向Be原子转移的现象.α相中P原子的0.07 e电子转移到Be原子,β相中P原子的0.12 e电子转移到Be原子,γ相中P原子的0.25 e电子转移到Be原子.
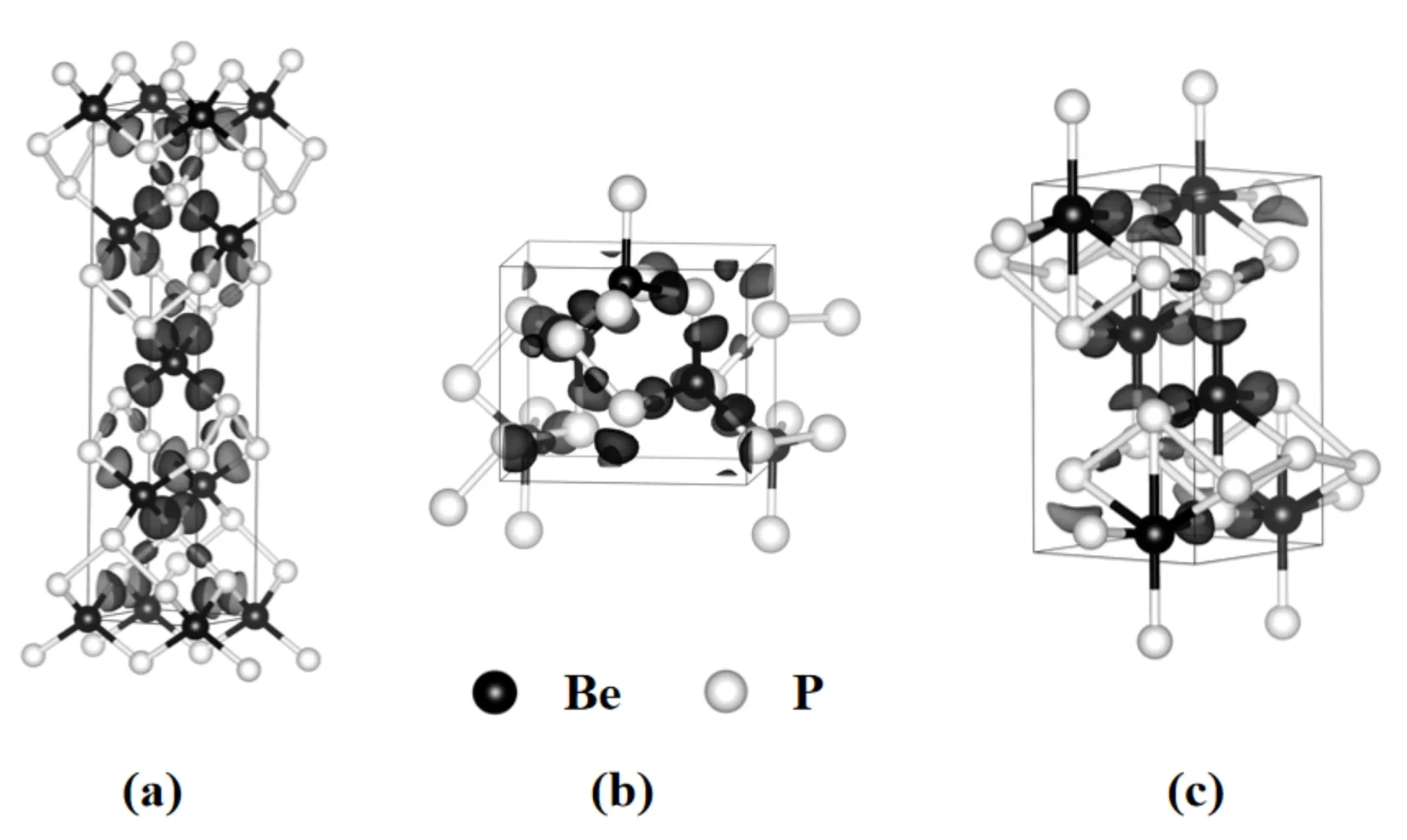
图5 α -BeP2相、β -BeP2相和γ -BeP2相的电子局域函数
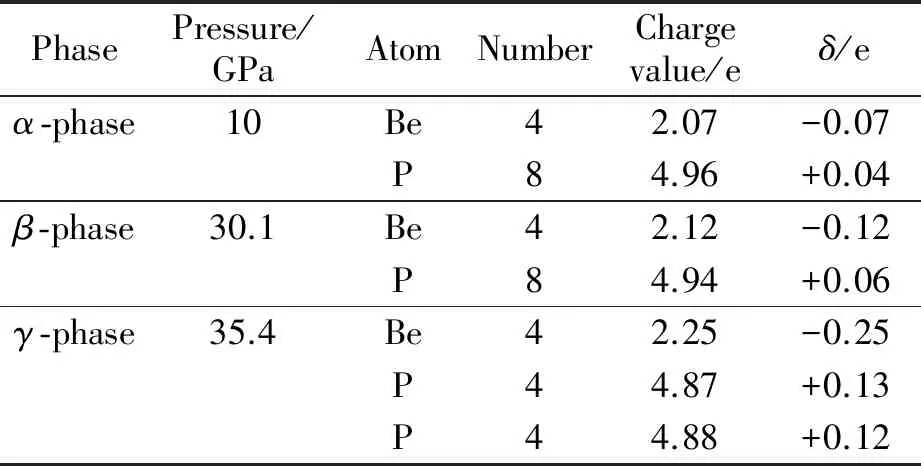
PhasePressure/GPaAtomNumberChargevalue/eδ/eα-phase10Be42.07-0.07P84.96+0.04β-phase30.1Be42.12-0.12P84.94+0.06γ-phase35.4Be42.25-0.25P44.87+0.13P44.88+0.12
3 结论
本文应用密度泛函理论和第一性原理赝势平面波方法,预测了高压下BeP2晶体的结构及其物理性质.预测结果表明:在常压下,α-BeP2相为立方结构,其空间群为I41/amd.当压强为30.1 GPa时,α-BeP2相发生结构相变,由α-BeP2相转变为β-BeP2相,其结构转变为四方结构,空间群为P43212.当压强为35.4 GPa时,β-BeP2相发生结构相变,由β-BeP2相转变为γ-BeP2相,其结构转变为正交结构,空间群为Imma.在上述预测结果中,常压相结构与实验结构完全吻合,高压相结构未被实验证实.电子性质计算发现,α-BeP2相和β-BeP2相的结构为窄带隙半导体,γ-BeP2相具有金属性质,这一结果为设计新型半导体材料提供了参考.计算高压下BeP2结构的电子局域函数和Bader电荷转移结果表明, P原子与P原子之间存在着共价键.
猜你喜欢
无机化学学报(2022年9期)2022-09-16
汽车实用技术(2022年5期)2022-04-02
小天使·聪聪画刊(2021年2期)2021-09-10
今日农业(2021年7期)2021-07-28
舰船科学技术(2021年12期)2021-03-29
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
汽车零部件(2020年10期)2020-11-09
中学生数理化(高中版.高考理化)(2020年2期)2020-04-21
汉语世界(The World of Chinese)(2019年6期)2019-09-10
汽车生活(2018年5期)2018-06-21
