
高效液相-质谱联用法测定改善睡眠类保健食品中的非法添加物
2019-05-24 00:55孙晓田仁芳董靖贾寒冰张亚锋
安徽医药 2019年6期
孙晓,田仁芳,董靖,贾寒冰,张亚锋
近年来,人们健康意识不断提升,对保健食品的需求也不断增加[1]。为了提高产品的市场竞争力,仍有人铤而走险将化学药品加入保健食品中,使保健食品产生速效作用,在短期内骗取群众口碑,进而增加销量谋取巨额利润[2]。其中,改善睡眠类保健食品需求量较大,临床上应用广泛的安神类化学药物成为了增强此类产品疗效的优先选择[3-4]。人们如果长期摄入这类化学药物,容易产生药物依赖性,因此对该类产品的风险监测迫在眉睫[5]。
目前保健食品中非法添加物的检测方法有薄层色谱法、紫外分光光度法、高效液相色谱法、高效液相-质谱联用法(HPLC-MS)等。其中薄层色谱法、紫外分光光度法操作简便,所需时间短,但专属性较弱[6]。高效液相色谱法具有灵敏度高、专属性好、设备费用适中的优点,但缺点是对于具有相同色谱行为,相同紫外吸收的同母核修饰化合物不具备分离鉴定能力[7-8]。本研究采用目前最先进科学的HPLC-MS/MS分析测试方法,于2016年7月开展研究,经大量方法学考察,结果证明该方法灵敏度最高、专属性最强。能较大程度避免假阳性结果的产生,同时能做到微量甚至痕量的准确分析,又能避免假阴性结果的产生[9],可推广应用于改善睡眠类保健食品中的非法添加物的检测监管,进而保障群众用药安全。
1 仪器与试药
1.1 仪器Agilent1200-6410B高效液相色谱-质谱联用仪(检测器三重四级杆QQQ,美国Agilent公司);电子分析天平(德国Mettler-Toledo系列);超声波清洗器(昆山市超声仪器有限公司)。
1.2 试药青藤碱(批号:110774-200507);佐匹克隆(批号:100870-200801);文法拉辛(批号:100543-200401);罗通定(批号:100452-200301),以上标准物质均购自中国药品生物制品检定院;氯苯那敏(中国食品药品检定研究院,批号:100047-200606);褪黑素(Dr.Ehrenstorfer.GmbH,批号:41222);扎来普隆(TRC,批号:4-ANR-96-1)。甲醇、冰醋酸(国药集团化学试剂有限公司,分析纯);乙酸铵(Aladdin,色谱纯)、乙腈(Merck,色谱纯);水(实验室自制,超纯水)。样品为本市辖区内随机抽样所得,001号为梦香源胶囊,002号为白色粉末,003号为安睡片。取阴性样品,加入上述混合标准物质适量,充分混合均匀,作为模拟阳性样品。
2 方法与结果
2.1 色谱-质谱条件色谱柱为CAPCELLPAK C18MG Ⅲ C18(150 mm×2.0 mm,5 μm)。流动相A为乙腈,流动相B为0.01 mol/L乙酸铵溶液(含0.1%乙酸),梯度洗脱:0~6 min,20%A;6~6.5 min,20%~40%A;6.5~15 min,40%~45%A;15~30 min,45%A;30~31 min,45%~20%A;31~36 min,20%A。流速0.3 mL/min;柱温35 ℃;进样量10 μL;检测器QQQ,离子源ESI+,雾化气温度300 ℃,雾化气流速10 L/min,MRM扫描,MRM母离子/子离子信息见表1[10-11]。
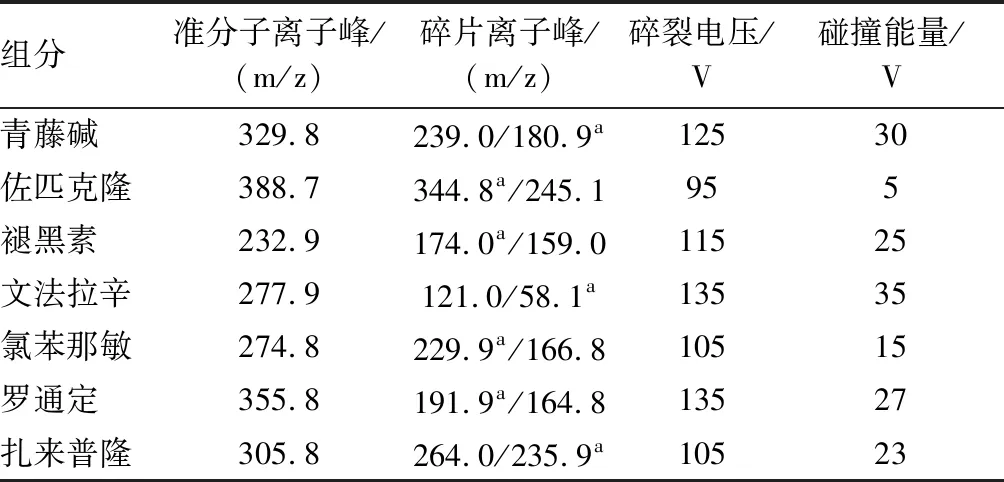
表1 化合物多反应监测离子信息
注:a为定量离子
2.2 标准物质溶液的配制分别精密称取青藤碱、佐匹克隆、褪黑素、文法拉辛、氯苯那敏、罗通定、扎来普隆标准物质各10.00 mg,分别置于25 mL容量瓶中,用甲醇溶解并定容至刻度摇匀,作为标准物质储备溶液;用刻度移液管分别精密移取以上标准物质储备溶液5 mL,置于50 mL容量瓶中,摇匀,作为混合标准物质溶液。
2.3 供试品溶液的制备取供试品5粒置于研钵中研磨均匀,精密称取0.45 g,于50 mL容量瓶中,加甲醇适量,超声处理15 min,取出放冷至室温,加甲醇定容至刻度,摇匀,备用。吸取上清液适量稀释500倍,0.22 μm有机系微孔滤膜滤过,取续滤液,即得。如果样品未检出非法添加物,建议再过滤一瓶未稀释的供试品母液,以排除假阴性结果。
2.4 专属性试验分别取阴性样品样品、模拟阳性样品按“2.3”项下处理,取混合标准物质、阴性样品、模拟阳性样品按“2.1”项下色谱条件进样测定,结果模拟阳性样品在与混合标准物质各组分相同保留时间处,离子流图一致。表明该方法可有效排除杂志干扰,专属性良好。总离子流图(TIC图)见图1。
2.5 标准工作曲线溶液的配制精密吸取“2.2”项下混合标准物质溶液适量,依次用甲醇稀释定容,制成混合标准工作曲线溶液,按照“2.1”项下色谱条件进样测定,以峰面积为纵坐标,浓度为横坐标绘制标准曲线,计算回归方程及相关系数r,结果表明各组分线性关系良好,见表2。
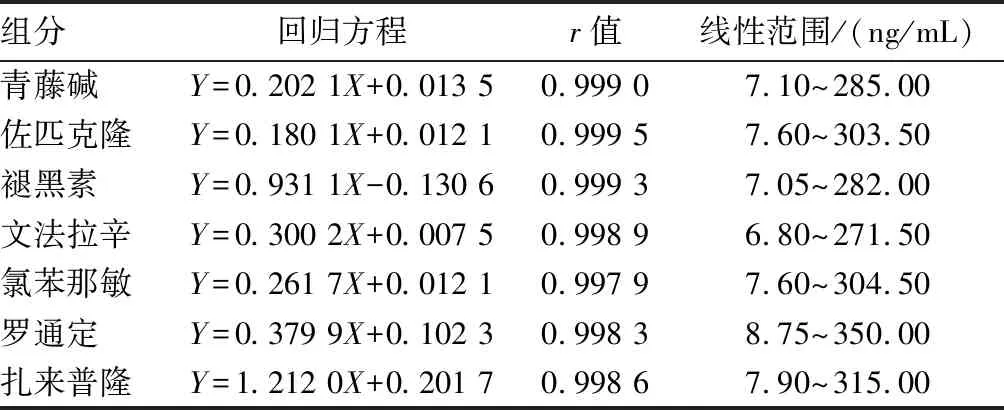
表2 标准曲线方程
2.6 精密度试验精密吸取混合标准物质溶液10 μL,按“2.1”项下色谱条件测定,连续进样6次,记录峰面积,分别计算7种组分的RSD(n=6)分别为1.37%,1.79%,1.01%,0.96%,1.51%,1.21%,0.86%,结果表明仪器精密度良好。
2.7 检出限试验将各组分标准物质逐级稀释,计算信噪比S/N=3时各组分浓度,结果确定青藤碱、佐匹克隆、褪黑素、文法拉辛、氯苯那敏、罗通定、扎来普隆检出限分别是1.78、2.17、1.76、1.70、1.90、2.19、1.58 ng/mL。
2.8 重复性试验精密称取模拟阳性样品6份,按照“2.3”项下方法同法处理,得到平行样品6份,按“2.1”项下色谱条件测定,记录峰面积,计算6份样品中7种组分的RSD分别为1.74%、1.91%、1.12%、0.99%、1.49%、0.97%、0.89%,结果表明该方法的重复性良好。
2.9 稳定性试验精密吸取模拟阳性样品溶液,分别在0、2、4、6、8、12 h按“2.1”项下色谱条件测定测定,记录峰面积,分别计算7种组分的RSD(n=6)分别为1.23%、1.01%、1.69%、0.91%、1.51%、1.03%、1.08%,表明样品12 h稳定性良好;精密标准物质溶液,分别在0、2、4、8、16、36 h按“2.1”项下色谱条件测定测定,记录峰面积,分别计算7种组分的RSD(n=6)分别为1.49%、1.17%、1.39%、1.27%、1.55%、1.26%、1.73%,表明标准物质12 h稳定性良好。
2.10 加标回收试验精密称取已知含量的样品9份,分为三组,每组3份,分别向每份样品加入混合标准物质溶液适量,制成高、中、低三梯度三平行的加标回收样品,按照“2.3”项下同法处理该9份样品,按“2.1”项下色谱条件测定,计算各组分回收率,结果表明该方法回收率可靠、稳定,见表3。
2.11 样品测定称取3批次保健食品样品,按照“2.3”项下方法处理,按“2.1”项下色谱条件测定,计算各组分含量,结果2批次未检出非法添加组分,1批次仅检出褪黑素,见表4。
3 讨论
本文所研究的检测方法,采用了更为先进、科学的HPLC-MS联用法。相比于传统的高效液相色谱法、薄层色谱法、理化鉴别等方法,能够极大程度上避免假阳性和假阴性结果的产生,灵敏度更高,定性/定量结果更准确,分析速度更快。
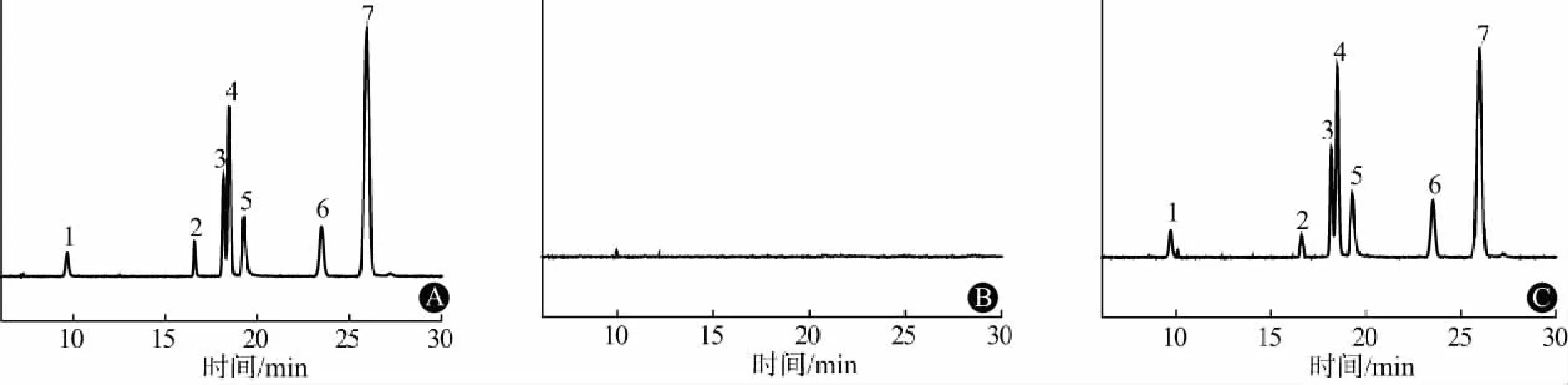
注:1.青藤碱;2.佐匹克隆;3.褪黑素;4.文法拉辛;5.氯苯那敏;6.罗通定;7.扎来普隆图1 总离子流图:A为混合标准物质;B为阴性样品;C为模拟阳性样品
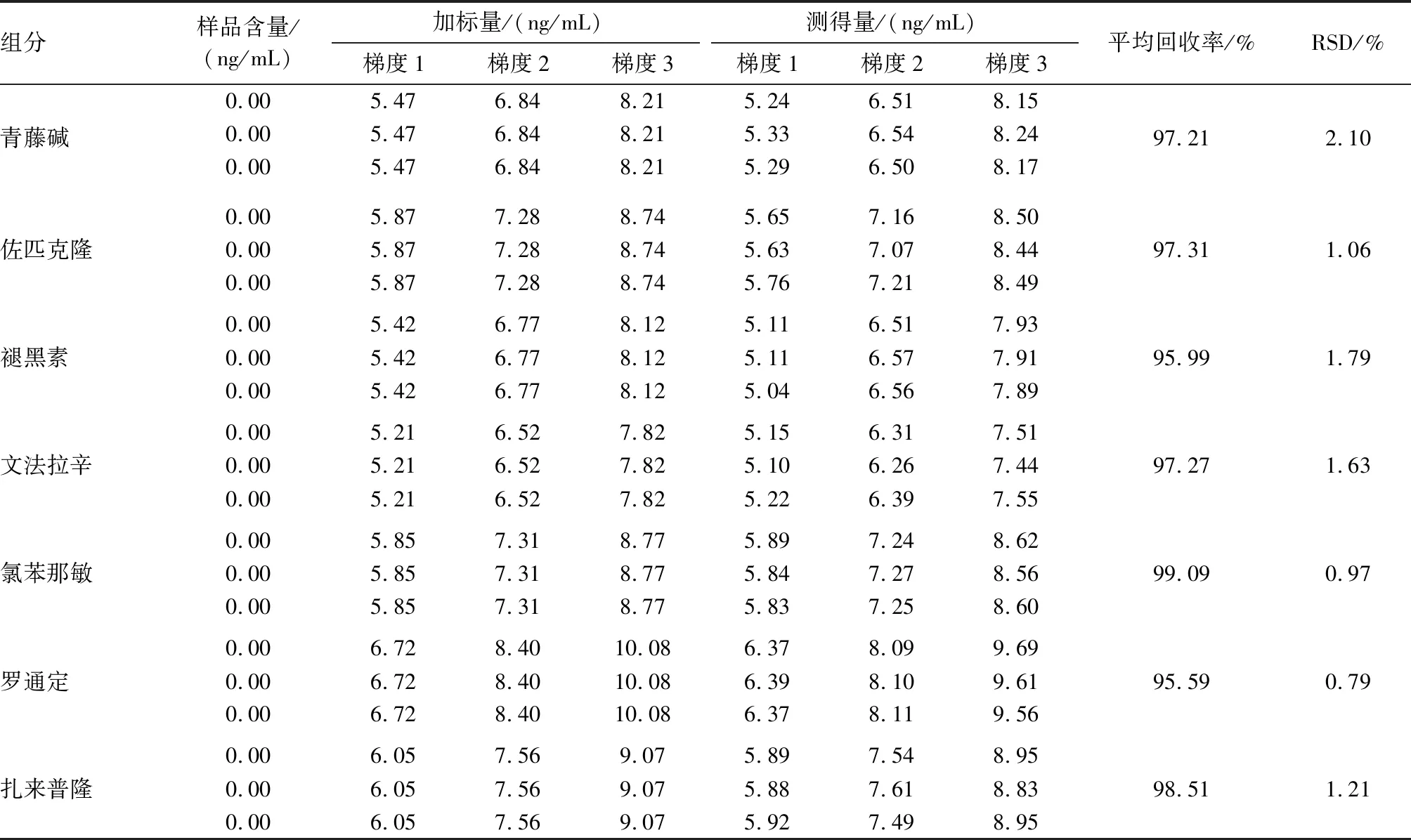
组分样品含量/(ng/mL)加标量/(ng/mL)测得量/(ng/mL)梯度1梯度2梯度3梯度1梯度2梯度3平均回收率/%RSD/%青藤碱0.005.476.848.215.246.518.150.005.476.848.215.336.548.240.005.476.848.215.296.508.1797.21 2.10佐匹克隆0.005.877.288.745.657.168.500.005.877.288.745.637.078.440.005.877.288.745.767.218.4997.31 1.06褪黑素0.005.426.778.125.116.517.930.005.426.778.125.116.577.910.005.426.778.125.046.567.8995.99 1.79文法拉辛0.005.216.527.825.156.317.510.005.216.527.825.106.267.440.005.216.527.825.226.397.5597.27 1.63氯苯那敏0.005.857.318.775.897.248.620.005.857.318.775.847.278.560.005.857.318.775.837.258.6099.09 0.97罗通定0.006.728.4010.086.378.099.690.006.728.4010.086.398.109.610.006.728.4010.086.378.119.5695.59 0.79扎来普隆0.006.057.569.075.897.548.950.006.057.569.075.887.618.830.006.057.569.075.927.498.9598.51 1.21
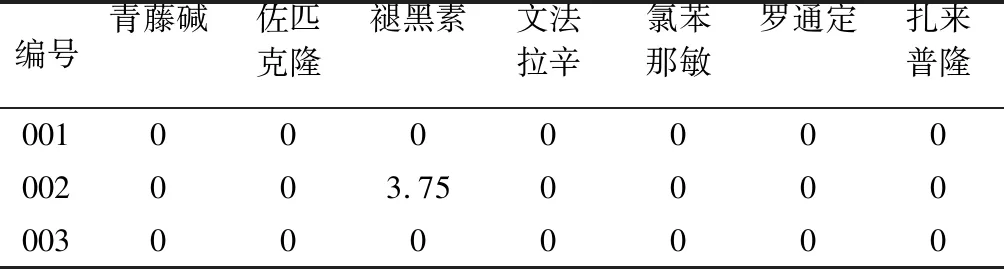
表4 样品测定结果/(g/kg)
将现有标准(编号2013002和编号2012004)中所囊括的7种安神类非法添加物质[12-13],由三种体系检测改进为单体系单针检测,效率更高结果更准确。相比于(编号BJS 201710)前处理过程,本研究方法增加了稀释步骤,可有效预防解决质谱检测器的污染问题;其次本方法针对改善睡眠类保健食品违禁药物添加问题,选择了针对性更强的化学添加药物进行检测,待测组分离子响应更强,干扰更小。
综上所述,本研究方法能够有效地缩短检测时间,节省实验人员、仪器、试剂等方面的成本,加快实验进度,提高实验准确度,提高了监管分析工作效率,具有很好的应用价值及前景。
猜你喜欢
航天电子对抗(2022年4期)2022-10-24
数学物理学报(2022年1期)2022-03-16
数学物理学报(2021年6期)2021-12-21
应用数学(2020年2期)2020-06-24
华东师范大学学报(自然科学版)(2019年3期)2019-06-24
中成药(2018年12期)2018-12-29
基层中医药(2018年7期)2018-12-06
中国科技纵横(2018年2期)2018-11-29
知识经济·中国直销(2018年11期)2018-11-26
中成药(2018年7期)2018-08-04
