
No evidence for an effect of Wolbachia on mtDNA variation and evolution in natural populations of Sesamia inferens (Lepidoptera: Noctuidae)
2019-05-10 06:13TANGXiaotianXUJingLUMingxingDUYuzhou
TANG Xiao-tian , XU Jing LU Ming-xing DU Yu-zhou
1 School of Horticulture and Plant Protection & Institute of Applied Entomology, Yangzhou University, Yangzhou 225009, P.R.China
2 Joint International Research Laboratory of Agriculture and Agri-Product Safety, Ministry of Education/Yangzhou University, Yangzhou 225009, P.R.China
3 Department of Entomology, Texas A&M University, TX 77840, USA
Abstract Wolbachia are widespread maternally-inherited endosymbiotic bacteria that infect numerous arthropods. This study represents a thorough survey of the Wolbachia infection patterns in the pink stem borer, Sesamia inferens (Walker), an important rice pest in China, based on nucleotide comparisons for the surface protein (wsp) and cell division protein (ftsZ) genes. The effects of Wolbachia on mtDNA variation and evolution of S. inferens were also investigated. Although we identif ied six genetically diverse strains, we found infections to be infrequent, with only 7.8% of hosts infected, and identif ied geographical differences in infection rates between southern and northern populations. Nucleotide indexes (haplotype diversity (Hd), nucleotide diversity (π) and number of variable sites (S) of mtDNA in infected populations were not signif icantly lower or higher than that in the uninfected populations. Furthermore, there was no association between Wolbachia infection status and phylogeny of mtDNA haplotypes. Analysis of molecular variance (AMOVA) showed that signif icant differentiation mainly existed within groups rather than among the groups. Additionally, using Tajima's D and Fu's F values, the mtDNA genes did not deviate signif icantly from neutral evolution. Taken together these results indicate that currently there were no effects of Wolbachia infection on host mtDNA variation and evolution in S. inferens.
Keywords: Wolbachia, Sesamia inferens, effect, mtDNA, prevalence
1. lntroduction
Wolbachia (Rickettsiae) are intracellular, maternally inherited bacteria known to infect a wide range of arthropods (Zhou et al. 1998; Cordaux et al. 2004). Across a variety of taxa, from 17 to 76% of tested insect species may be infected with Wolbachia (Werren et al. 1995a; Wenseleers et al. 1998; Jeyaprakash and Hoy 2000; Kittayapong et al. 2000a, b; Werren and Windsor 2000; Hilgenboecker et al. 2008). Recent surveys indicate that the genus Wolbachia comprises 17 ‘‘supergroups'' (A to Q) (Baldo and Werren 2007; Augustinos et al. 2011; Jiang et al. 2014; Glowska et al. 2015). Supergroups A and B are most commonly found among arthropod species (Werren et al. 1995b; Lo et al. 2007; Duron et al. 2008). There is a long-term co-evolutionary relationship between Wolbachia and their hosts for thousands (or millions) of years, and it has been determined that Wolbachia can cause a number of reproductive alterations in their hosts, including the induction of cytoplasmic incompatibility (CI), thelytoky (T) or feminization (F) (Legrand et al. 1980; Rousset et al. 1992; Juchault et al. 1994; Field et al. 1999; Pintureau et al. 2000; Pannebakker et al. 2004; Blagrove et al. 2012; Rabeling and Kronauer 2013; Valette et al. 2013; Kern et al. 2015; Richardson et al. 2016; Cooper et al. 2017). In addition, there exists a linkage disequilibrium between Wolbachia and host mitochondrial DNA as they are both co-transmitted maternally (Jiang et al. 2014). Numerous studies have found that Wolbachia can potentially inf luence the mitochondrial variation as well as the evolution of their hosts (Marcade et al. 1999; Dean et al. 2003; Dewayne et al. 2003; Dyer and Jaenike 2004; Hurst and Jiggins 2005; Galtier et al. 2009; Yu et al. 2011; Zhang et al. 2013a; Chen et al. 2016; Klopfstein et al. 2016; Cariou et al. 2017). This inf luence is exhibited in several ways. For example, infected and uninfected individuals have been found to exhibit divergent mtDNA sequences (Marcade et al. 1999); other studies have found reduced or increased mtDNA diversity in infected rather than uninfected species (Dean et al. 2003; Jiggins 2003; Dyer and Jaenike 2004; Yu et al. 2011; Zhang et al. 2013a). Still others have found Wolbachia to be associated with a subset of mitotypes in the population (Grandjean et al. 1993; Rigaud et al. 1999).
The pink stem borer, Sesamia inferens (Walker) (Lepidoptera: Noctuidae), is an important pest of rice in China and other Asian countries (Sun et al. 2014; Tang et al. 2014). With the expansion of hybrid rice plantings as well as changes in climate and rice cultivation systems, the S. inferens population has been gradually increasing in China, and has become an important pest alongside Chilo suppressalis (Walker) since 1990 (Sheng et al. 2003). Although Wolbachia have been detected in numerous rice pests (Kittayapong et al. 2003; Chai et al. 2011; Chai and Du 2011; Chen et al. 2012), little is known about their distribution in S. inferens. Because of the limited number of genetic studies, little is known about the distribution of Wolbachia in S. inferens or whether Wolbachia affect mtDNA of S. inferens or not, which limited the application of mtDNA markers in population genetics research of S. inferens.
The Wolbachia surface protein gene (wsp), cell division protein gene (ftsZ), 16S rRNA, and other genes have been characterized and used for phylogenetic studies of this endosymbiont. The wsp and ftsZ genes are usually considered to be faster evolving than 16S rRNA, with about 10 times more variable than the bacterial 16S rDNA gene (Zhou et al. 1998). These two genes have been used for supergroup designation and as molecular markers in the study of the phyletic evolution of Wolbachia (Zhou et al. 1998; Haine and Cook 2005; Kyei-Poku et al. 2005; Sintupachee et al. 2006; Park et al. 2016; Vivero et al. 2017); meanwhile, in most cases, the wsp gene is unsuited for strain designation or phylogeny reconstruction due to its high level of recombination (Baldo et al. 2006; Baldo and Werren 2007); therefore, we only use it as complement in this study.
The objectives of this study were to: 1) investigate the Wolbachia infection status of S. inferens in China based on wsp and ftsZ genes; and 2) explore the potential effects of Wolbachia on the variation and evolution of mtDNA in S. inferens. This work provides data to supplement other studies of molecular diversity of Wolbachia in Lepidoptera (Ilinsky and Kosterin 2017) and inform future population genetic diversity studies of S. inferens.
2. Materials and methods
2.1. Samples collection and DNA extraction
S. inferens samples were collected in rice f ields during 2013-2014 throughout China. Our 27 sampling locations included the most important locations for rice production (Mei et al. 1988; Duan and Zhou 2011) and S. inferens infestation (Xu et al. 2011) (Table 1). Individuals were collected as larvae from infested rice stems and the larvae were preserved in 100% ethanol at -20°C. Genomic DNA was extracted from S. inferens larvae using an AxyPrep Multisource Genomic DNA Miniprep Kit (Axygen, Suzhou, China) as recommended by the manufacturer.
2.2. Detection of Wolbachia and amplif ication of host mtDNA genes
The primers wsp81F and wsp691R were used in the PCR to amplify the wsp fragments from samples as described by Zhou et al. (1998). The Wolbachia-specif ic primers ftsZ f1 and ftsZ r1 were used for amplif ication of about 1 000 bp of the Wolbachia cell division protein gene ftsZ (Werren et al. 1995b). Amplif ication reactions were run in 50 µL buffer using the TaKaRa Taq Kit (TaKaRa, Japan): 36.5 µL H2O, 3.5 µL 10× buffer, 4 µL of 2.5 mmol L-1dNTPs, 1.5 µL of 25 mmol L-1MgCl2, 0.5 µL r Taq polymerase (5 U µL-1, TaKaRa), 2 µL sample and 1 µL primers (10 mmol L-1each). Cycling conditions were 94°C for 5 min followed by 35 cycles of 94°C for 30 s, Tm (53°C for wsp; 55°C for ftsZ) for 30 s and 72°C for 1 min and f inally 72°C for 10 min. Each sample had three replicates to ensure detection accuracy.
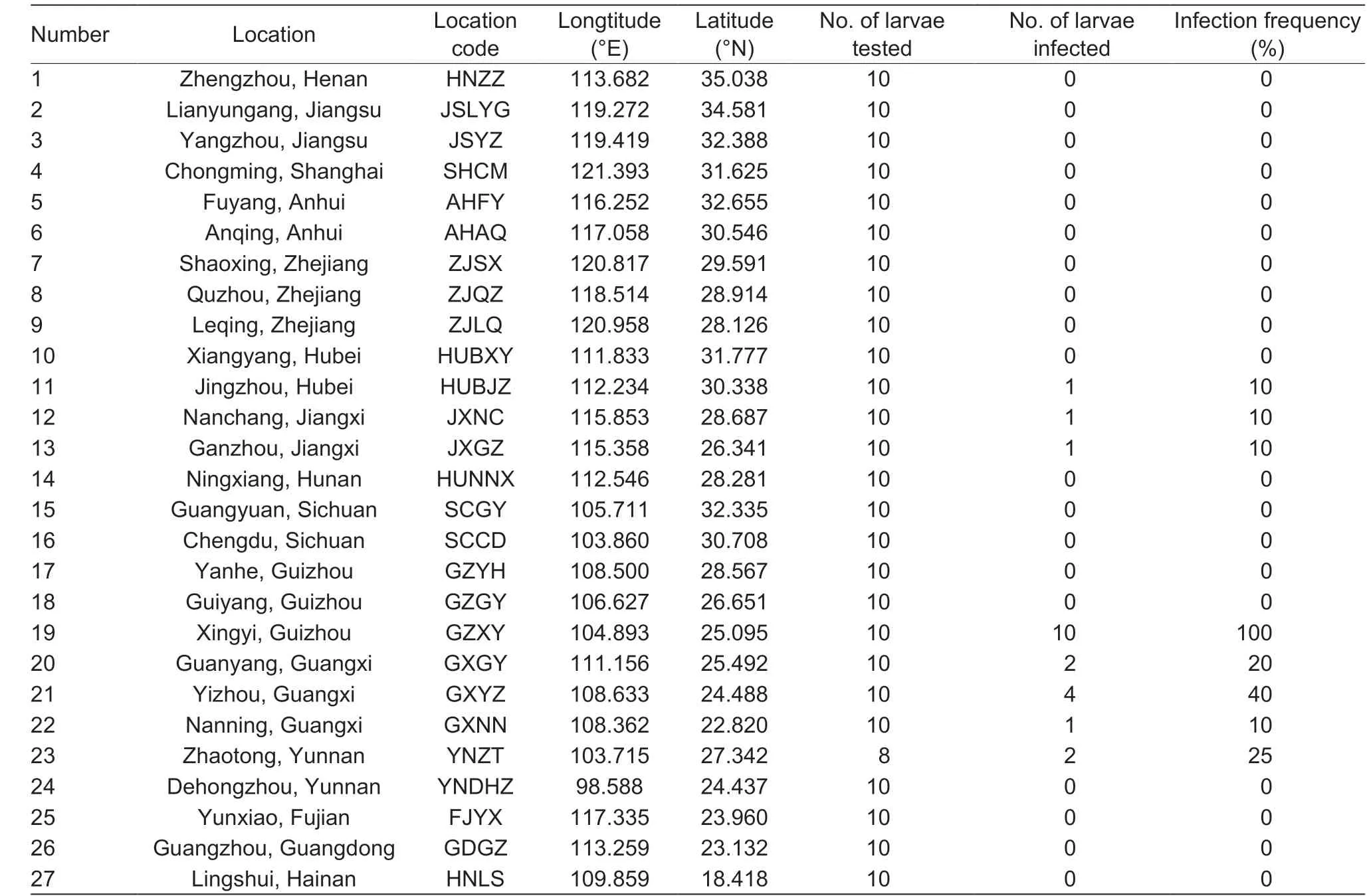
Table 1 Collection site, code, number of studied individuals and Wolbachia infection rate in Sesamia inferens
Portions of two mitochondrial genes (Cytochrome oxidase subunit I (COI) and cytochrome b (Cytb)) from infected populations were amplif ied from each individual using the primer pairs: LeCOI-F (5´-GGAGCAGGAACTGGATGA-3´) with Le COI-R (5´-GCAGGGGGTAGATTTTGA-3´), which obtained a 976-bp fragment of COI, and CP1 (5´-GATGATGAAATTTTGGATC-3´) (modif ied from Harry et al. 1998) with Tser (5´-TATTTCTTTATTATGTTTTCAAAAC-3´) (Simon et al. 1994), which amplif ied a 998-bp fragment of Cytb. PCR reactions were carried out in 50 µL reaction aliquots containing 100 ng extracted DNA, 5 µL 10× buffer, 4 µL 25 mmol L-1MgCl2, 4 µL 2.5 mmol L-1d NTPs, 1 µL 20 µmol L-1primers, 0.4 µL Taq DNA polymerase (5 U µL-1) and ultra-pure water. PCR of the reaction mixtures was done with the following temperature prof ile for the gene fragment: 94°C for 5 min; 35 cycles of 94°C for 1 min and 15 s, Tm (53°C for COI and 48.9°C for Cytb) for 1 min and 15 s, 72°C for 1 min and 5 s, and a f inal extension step of 72°C for 10 min.
All amplif ication products above were purif ied using the AxyPrep™ DNA Gel Extraction Kit (Axygen, Union City, CA, USA), cloned into p GEM-T Easy vector (Promega, USA), and then transformed into Escherichia coli DH5α cells. Positive clones were picked and sequenced by IGE Biotechnology Co., Ltd. (Guangzhou, China).
2.3. Data analysis
The GenBank database was searched for homologous sequences of wsp, ftsZ, COI and Cytb genes using the Basic Local Alignment Search Tool (BLAST). Sequences were initially aligned using the CLUSTAL X 1.83 Program (Chenna et al. 2003) and manually ref ined. Phylogenetic trees were constructed using MrBayes ver. 3.2.1 (Ronquist and Huelsenbeck 2003) with Anaplasma marginale and Anaplasma Phagocytophilum as outgroups. The phylogenetic status of Wolbachia from S. inferens was investigated by comparing our sequences to 40 wsp gene sequences (Zhou et al. 1998; Van Meer et al. 1999) and 26 ftsZ sequences (Casiraghi et al. 2005) from NCBI. The best f it model for nucleotide alignments was determined by Modeltest 3.7 (Posada and Buckley 2004). According to the Akaike information criterion, the GTR+I+G paradigm was the best model for analysis using nucleotide alignments. The nucleotide alignments were constructed using the MrBayes Program with 1 100 000 generations and with the f irst 25% discarded as burn-in. Tree information was visualized and edited using FigTree ver. 1.3.1 (Rambaut 2009). Median-joining networks of haplotypes of the two genes were constructed using NETWORK v. 4.6 (Bandelt et al. 1999). Phylogenetic signal estimates the degree to which interspecif ic trait variation is correlated with phylogenetic relationships (Haak et al. 2014). Therefore, we used the Blomberg's K statistic (Blomberg et al. 2003) as implemented in the phylosignal function in the picante package (Kembel et al. 2010) to evaluate whether traits of Wolbachia strains were signif icantly associated with mitochondrial phylogenetic relationships. We also investigated correlation between the infection rate and latitude, altitude, temperature (max. and min.) as well as precipitation.
To test whether selective sweeps by Wolbachia infection affected host mtDNA variation or evolution, we compared nucleotide indexes among uninfected and infected individuals, and mtDNA haplotype diversity (H), nucleotide diversity (π) and the number of variable sites (S) were calculated in DnaSP 5.0 (Librado and Rozas 2009). Genetic differentiation was investigated by AMOVA (analysis of molecular variance) and the related f ixation indices (FST) as implemented in the program Arlequin 3.5 (Excoff ier and Lischer 2010). Tajima's D and Fu's F statistics as well as mismatch distributions analysis were also calculated using Arlequin 3.5 to test the neutral mutation hypothesis.
3. Results
3.1. Wolbachia infection in S. inferens populations
A total of 268 f ield-collected S. inferens larvae across China were tested. We found 7.8% of all specimens to be Wolbachia-positive based on detection of the two proteincoding genes (wsp and ftsZ). The lengths of wsp and ftsZ sequences amplif ied was 598-619 and 1 045-1 057 bp, respectively. In total, 13 wsp and seven ftsZ haplotypes were identif ied in this study that had not been reported previously. All of the haplotypes have been deposited in the GenBank database (KP822797-KP822809; KP844447-KP844453; Appendix A).
Eight populations (of 27) were infected with Wolbachia, with the highest infection rate of 100% in the GZXY population and the lowest infection rate of 10% in several other populations (e.g., HUBJZ and JXGZ) (Table 1 and Fig. 1) and we found that Wolbachia infection was more prevalent in borer populations in southern China, especially in Guangxi, Guizhou and Yunnan (Fig. 1 and Table 1). This probably because the infection rate has correlation with altitude (r2=0.26275), precipitation (r2=0.21972) and maximum temperature (r2=0.33527) to some extent based on our correlation analysis. Taken together, these results suggest infection by Wolbachia in S. inferens is polymorphic but relatively rare in China, especially in northern regions.
3.2. Wolbachia phylogeny
To explore the phylogenetic status of Wolbachia from S. inferens, 21 partial sequences of the wsp gene amplif ied from S. inferens together with 40 wsp sequences described in previous research (Zhou et al. 1998; Van Meer et al. 1999), and 14 ftsZ sequences coupled with 26 sequences from Casiraghi et al. (2005) were analyzed using the Bayesian inference. The resulting topology shows that 14 (of the 21) wsp sequences belong to supergroup B, and the remaining seven to supergroup A (Fig. 2). Using the def inition of Van Meer et al. (1999) and Zhou et al. (1998), we were able to def ine the six strains of Wolbachia from S. inferens, including four common strains (w Dro, w Ori, w Pip and w Con) and two new strains (w Cam and w Inf) proposed in this study (Fig. 2). In detail, JXNC6, one of the two wsp sequences of the above new strains, was largely similar to the Wolbachia sequence from Spalangia cameroni Perkins (Hymenoptera: Pteromalidae) (Kyei-Poku et al. 2006) with the sequence differences of 0.66%, and we proposed it to be a new strain and we named “w Cam”. Another wsp sequence, GZXY9, was very different from those already submitted in GenBank, and we proposed it as a new strain we named “w Inf” according to the species name.
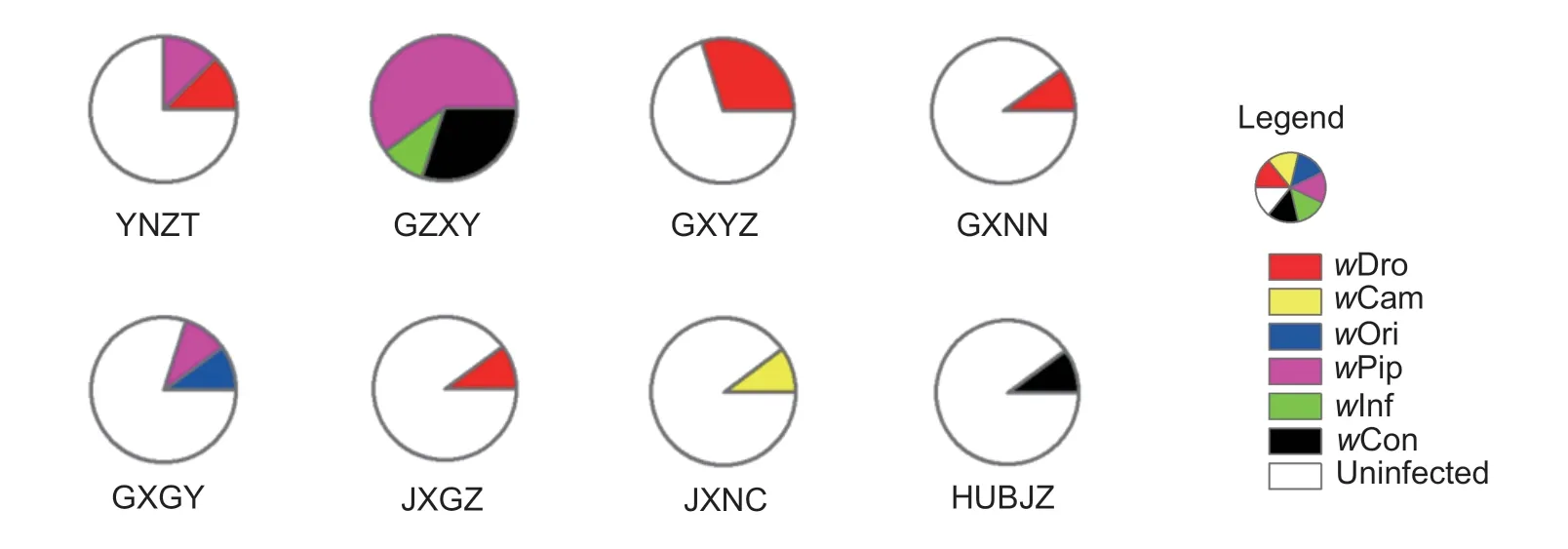
Fig. 1 Wolbachia infection status and strain types in eight infected populations. w Dro, w Cam, w Ori, w Pip, w Inf and w Con are strains of Wolbachia. Strain identif ications are based on Zhou et al. (1998) and Van Meer et al. (1999).
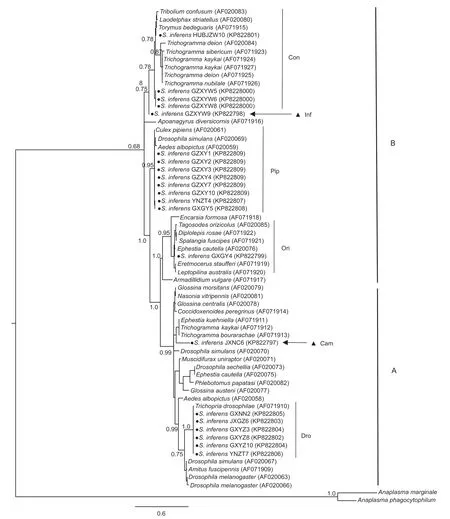
Fig. 2 Bayesian inference phylogeny of Wolbachia based on wsp gene sequences. Posterior probability values exceeding 50% (1 000 replicates) are indicated. Names at the terminal nodes are those of the host species. Accession numbers are given for the sequences present in the GenBank databases. Strain identif ications (such as w Dro and w Ori) are according to Zhou et al. (1998) and Van Meer et al. (1999). Anaplasma marginale and Anaplasma phagocytophilum are outgroups. A and B denote two of the Wolbachia supergroups. Bold dots indicate wsp gene sequences from Sesamia inferens. Triangles indicate two newly proposed strains of Wolbachia.
There is variation of the Wolbachia strains within the populations we studied. Four of the infected populations (JXGZ, GXNN, GXYZ and YNZT) were infected with strain w Dro, which was the most common Wolbachia strain we found (Fig. 1). Strain w Pip was also common in three populations (GXGY, GZXY and YNZT), whereas strains w Cam, w Ori and w Inf each infected individual in only one population. In contrast, the GXGY, GZXY and YNZT populations were each infected by at least two strains of Wolbachia, while the remaining f ive populations were each infected by only one strain (Fig. 1).
In addition, the ftsZ gene sequences could be divided into A and B supergroups with 11 sequences in the supergroup B and three in the supergroup A (Fig. 3). The Wolbachia sequences in the GXYZ population were all clustered into the A supergroup, which is consistent with results of the wsp gene (Fig. 2). Furthermore, we identif ied three strains (I, II and III) based on ftsZ sequences (Fig. 3).
3.3. No effects of Wolbachia on mtDNA of S. inferens
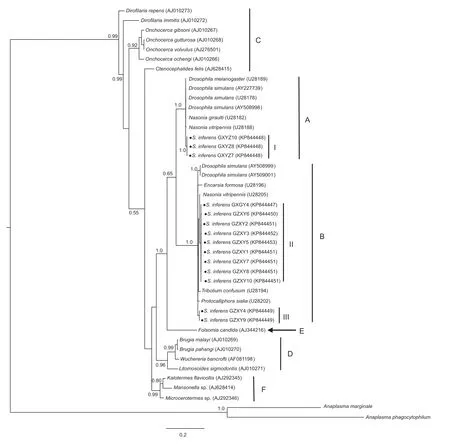
Fig. 3 Bayesian inference phylogeny of Wolbachia based on ftsZ gene sequences. Posterior probability values exceeding 50% (1 000 replicates) are indicated. Names at the terminal nodes are those of the host species. Accession numbers are given for the sequences present in the GenBank databases. Anaplasma marginale and Anaplasma phagocytophilum are outgroups. Supergroups A-F are depicted and named according to Casiraghi et al. (2005). Bold dots indicate ftsZ gene sequences from Sesamia inferens. I, II and III are three Wolbachia strains.
As mitochondrial DNA is in linkage disequilibrium with the maternally inherited bacteria, Wolbachia could affect mitochondrial DNA variation and evolution (Baudry et al. 2003; Jiggins 2003; Charlat et al. 2009). To explore whether Wolbachia from S. inferens affect its mtDNA variation and evolution or not, partial COI and Cytb gene sequences were obtained from Wolbachia-infected populations. In total, we identif ied 36 haplotypes for concatenated sequences (COI+Cytb) and 28 haplotypes for both COI and Cytb sequences, respectively (Table 2; Appendix B). Moreover, 26 haplotypes in uninfected individuals while 14 haplotypes of concatenated sequences were observed in infected individuals (Table 2). Importantly, we found that Hd, π and S in infected populations were not signif icantly lower or higher than that in the uninfected group, with values close to 0.9, 0.03, and 250, respectively (Table 2). The analysis based on individual gene showed the similar pattern (Table 2). It is noteworthy that the mtDNA haplotypes were not concordant with the strains of Wolbachia. For example, Hap COI15, Hap COI21, Hap COI24, Hap COI26 and Hap COI28 only have the strain w Dro, and HapCOI16 shared by three strains (w Pip, w Inf and w Con) (Fig. 4-A). Cytb haplotype data exhibited the similar characteristics (Fig. 4-B). The network based on ftsZ strains showed the similar pattern (Appendix C). This observation was also supported by our analyses of phylogenetic signal in ftsZ strain traits. The Blomberg' s K value (K=0.078, P=0.001) was extremely low and signif icant, indicating little phylogenetic signal in strain traits. We also found there was no association between Wolbachia infection status (infected or uninfected) and phylogeny of mtDNA haplotypes (Fig. 4). This suggested that the mtDNA of infected and uninfected S. inferens individuals has not evolved independently, and the haplotypes of infected individuals were not clustered into the same lineage. For example, HapCOI13 and Hap Cytb7 were detected in both infected and uninfected individuals.
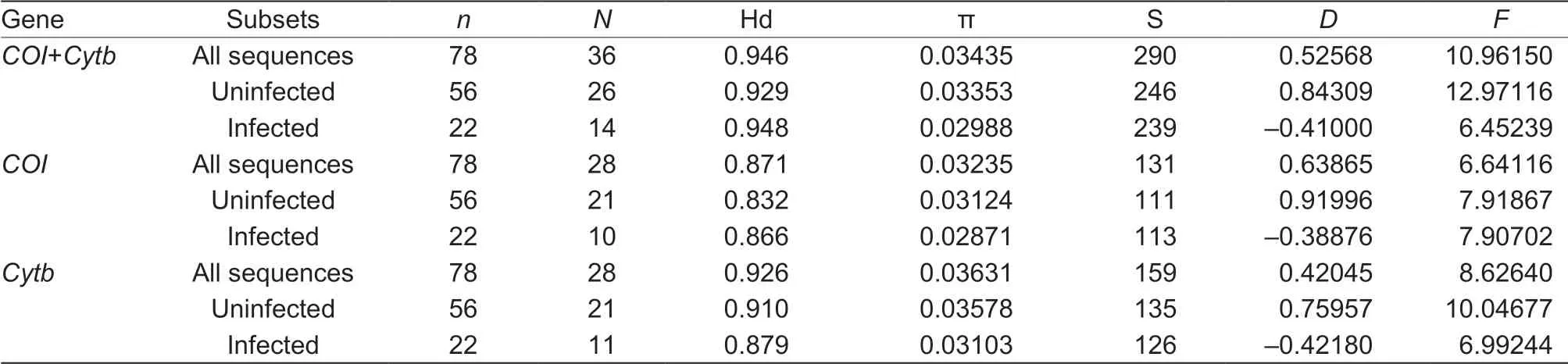
Table 2 Nucleotide polymorphisms of mtDNA genes in infected and uninfected Sesamia inferens samples1)
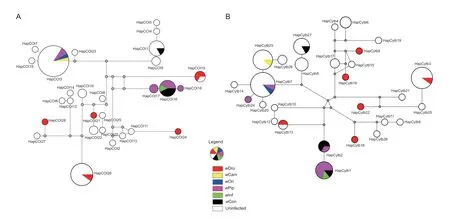
Fig. 4 Median-joining network of Sesamia inferens mtDNA (COI, A) and mtDNA (Cytb, B) haplotypes associated with Wolbachia infection status based on wsp gene. The area within the circles are proportional to haplotype frequency.
Analysis of molecular variance (AMOVA) clearly shows that signif icant differentiation (COI+Cytb, FST=0.12051, P<0001; COI, FST=0.12642, P<0.001; Cytb, FST=0.11532, P<0.0001) existed within groups (def ined by infected and uninfected) (87.95, 87.36 and 88.47%, respectively) rather than among groups (12.05, 12.64 and 11.53%, respectively) (Table 3). This indicates that the Wolbachia status has no effect on the variation of mtDNA in S. inferens.
Positive values for Tajima's D and Fu's F statistics indicate an excess of intermediate-frequency variants, whereas signif icant negative values indicate an excess of rare variants, which can result from a recent population bottleneck or processes such as background selection (Yu et al. 2011). In our study, estimates of Tajima's D were generally negative but not signif icant for the Wolbachiainfected group (Table 2), indicating that selective sweeps by Wolbachia infection have not affected mtDNA diversity. Moreover, we also detected selective sweep by mismatch analysis, which could be used to test hypotheses about the history of population size and subdivision or about selection (Rogers and Harpending 1992; Rogers et al. 1996). According to Rogers and Harpending (1992), the observed curves with numerous peaks or resemblance to expected curves mean equilibrium population and unimodal curves representing population expansion. In our study, the observed mismatch analysis exhibits a multimodal distribution of pairwise differences for the infected and uninfected group (Fig. 5; Appendix D), indicating equilibrium in the S. inferens populations for both the Wolbachia infected and uninfected groups. However, the genetic differentiation of mtDNA does appear to be related to the presence of different Wolbachia strains. Pairwise FSTvalues showed signif icant genetic differentiation between the uninfected and the infected group for w Pip and w Con strains based on COI, Cytb and concatenated sequences (Table 4; Appendices E and F). Furthermore, all of the three strains based on ftsZ gene showed signif icant FSTvalues compared to uninfected group (Appendix G).
4. Discussion
Our results indicate a very low prevalence (7.8%) of Wolbachia infection in S. inferens. This is consistent with one study of Pieris rapae, in which Wolbachia was detected with a prevalence of 3.4% (Tagami and Miura 2004). One explanation for this, proposed by Hamm et al. (2014), is that with no apparent host reproductive manipulation, Wolbachia exhibit imperfect maternal transmission, which systematically reduces its frequencies. It is also possible that the extent of Wolbachia infection is inf luenced by the climate or the environment (Werren and Windsor 2000; Keller et al. 2004). Thus, temperature or other climate factors may restrict Wolbachia infections, especially in northern China populations of S. inferens. Technically, the low detection could be due to the two selected genes (wsp and ftsZ). For example, Augustinos et al. (2011) reported successful 16S rRNA amplif ication in 35 aphids samples, whereas amplif ication of the ftsZ gene was successful in only two samples. Therefore, more proper genes should be tested in future research.
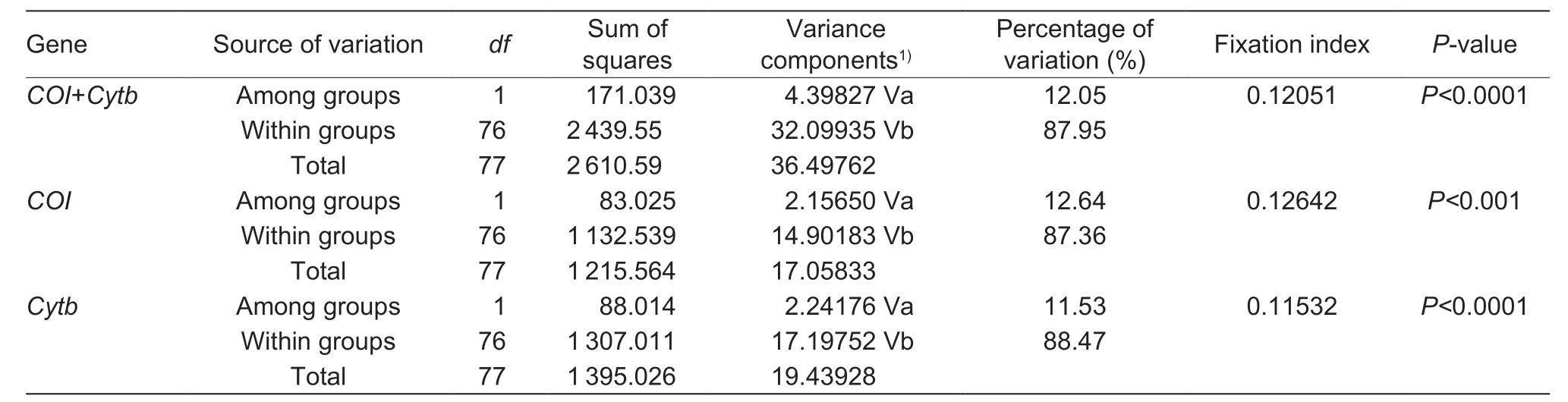
Table 3 Analysis of molecular variance (AMOVA) comparing genetic variation in infected and uninfected Sesamia inferens groups
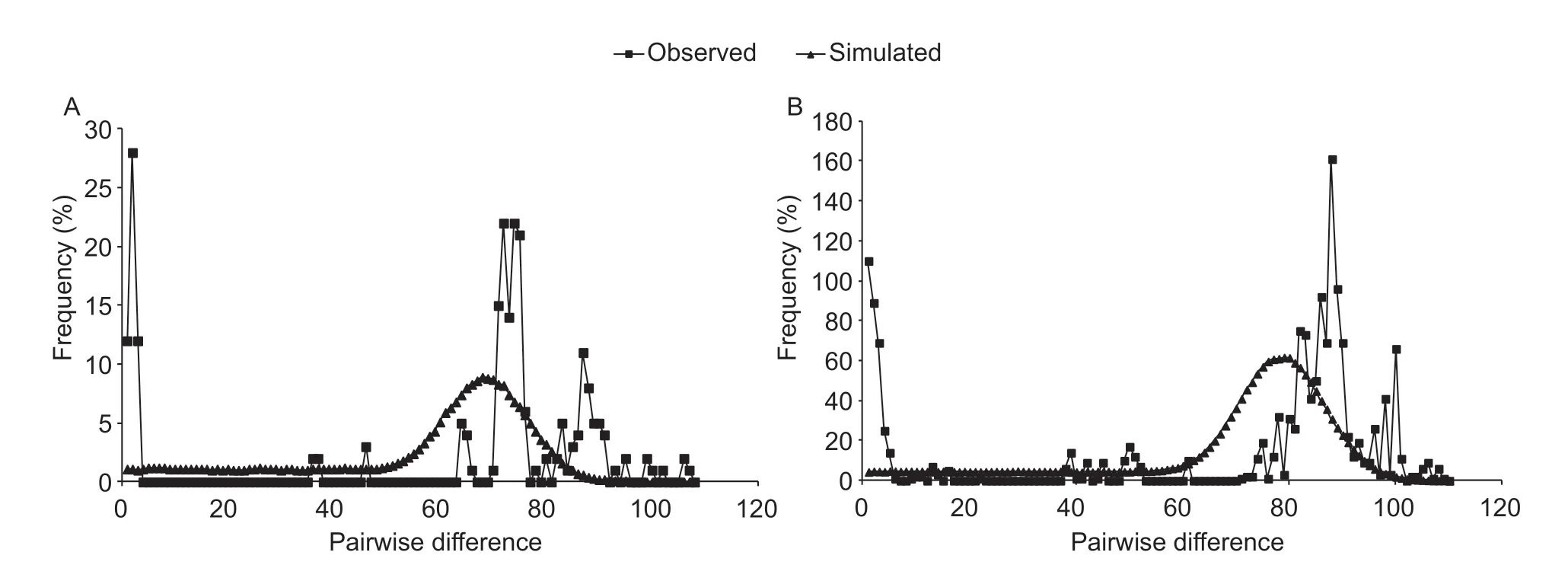
Fig. 5 Observed and simulated mismatch distributions of Sesamia inferens. A, infected group based on concatenated sequences (COI+Cytb). B, uninfected group based on concatenated sequences (COI+Cytb).
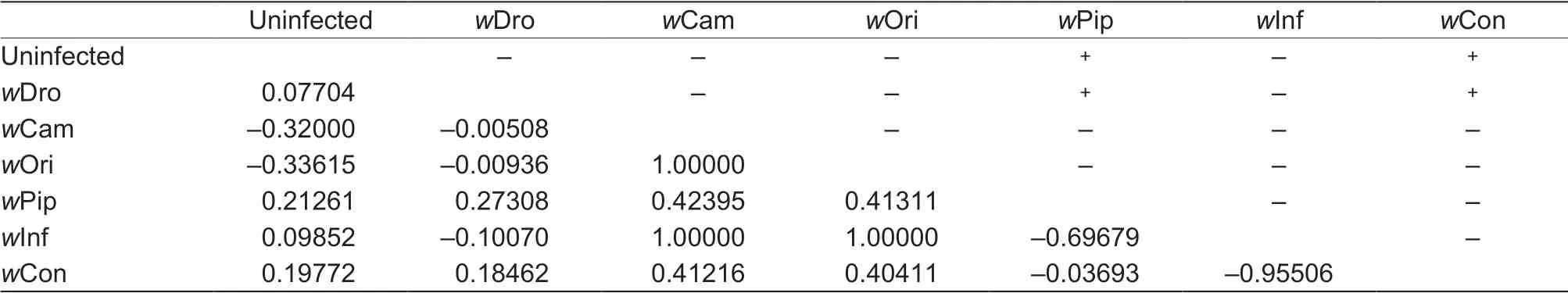
Table 4 Fixation index (F ST) values of concatenated sequences (COI+Cytb) inferred from pairwise-compared wsp strains
Despite a low prevalence of Wolbachia in S. inferens, various Wolbachia strains were identif ied in the current study. It is not well understood what factors are responsible for variations in the number of Wolbachia strains coinfecting host species (Reuter and Keller 2003). The multiple Wolbachia strains might be related to the habitats environment of S. inferens or horizontal transmission of Wolbachia. It has been proposed that some hosts might be less exposed to environmental curing and therefore harbor more strains because they lose infections at a lower rate (Jamnongluk et al. 2002). Alternatively, for horizontal transmission, parasitoid wasps might have to be considered as a causal factor and the evidence showed that Wolbachia can use parasitoid wasps as phoretic vectors for eff icient horizontal transmission (Ahmed et al. 2015). When host eggs are parasitized by parasitoids, the Wolbachia hosted by the latter would come into host eggs and thereafter potentially become a “novel” strain in the host. Furthermore, horizontal transmission of Wolbachia possibly facilitates the invasion of new strains which could be given the evidence that the most common Wolbachia strain w Dro was widely found in S. inferens-infected populations. Besides, high levels of multiple infection may also stem from non-migratory character of S. inferens. A contrasting example is from Cnaphalocrocis medinalis (Guenée) (Chai et al. 2011), a migratory pest of rice. C. medinalis have been found that the ftsZ and 16S rDNA sequences were exactly the same in all positive samples from different regions, which might be due to the high level of migration causing the same strain of Wolbachia to be widely transmitted to different populations (Chai et al. 2011).
Our results of Wolbachia prevalence varied between climatic regions. We found Wolbachia was absent from individuals in the northeast populations, while southwest populations were relatively highly infected with Wolbachia, especially in Guangxi. Similar patterns of distribution have been reported for several host-insect species. For example, in another Lepidopteran insect, Polytremis nascens Leech, individuals from southern China are highly infected with Wolbachia (Jiang et al. 2014). The incidence of Wolbachia of tephritid fruit f lies was higher in the northern half of the gradient while none was infected in the southern half in Australia (Morrow et al. 2015). It is interesting that a contrary example, Wolbachia diversity of Tetranychus truncates Ehara (Acarina: Tetranychidae) appeared to be higher in north region (Zhang et al. 2013b). Anyway, an increasing number of studies have demonstrated that Wolbachia responds to the climatic environment of its host insects (Werren and Windsor 2000; Keller et al. 2004). It is possible that temperature f luctuations and extremes in northern regions of China may provide more selective pressure towards Wolbachia of S. inferens than that in temperate regions. This may explain the 100% Wolbachia infection rate reported here for the GZXY population, where temperature, humidity or altitude may favor the reproduction and spread of Wolbachia as we indicated the correlations above.
Due to the fast evolution of Wolbachia and especially the high frequency of recombination events, it might be misleading to classify Wolbachia strains based only on wsp sequences (Baldo et al. 2005; Baldo and Werren 2007; Yang et al. 2012). We therefore used the ftsZ gene from multilocus sequence typing (MLST) to verify Wolbachia phylogeny based on the wsp gene. It is noteworthy that Wolbachia sequences in GXYZ population were still all clustered into the A supergroup (Fig. 3). Moreover, we found that most Wolbachia sequences were in the B supergroup whether based on the wsp or on the ftsZ gene (Figs. 2 and 3). This is consistent with Werren and Windsor (2000), who found Hymenoptera to be more frequently infected with the A supergroup of Wolbachia while Lepidoptera to be more frequently infected with the B supergroup. Our results suggest that Wolbachia phylogenies based on wsp and ftsZ genes are largely similar, strengthening accuracy conf idence in the phylogeny results.
There are numerous reports of problems with using mtDNA as a marker in population genetics, phylogeographic and phylogenetic studies due to the effects of inherited symbionts (Hurst and Jiggins 2005; Galtier et al. 2009; Klopfstein et al. 2016; Schuler et al. 2016). Decreased mitochondrial DNA polymorphism as a consequence of Wolbachia infection has been reported in numerous studies (e.g., Shoemaker et al. 1999, 2004; Baudry et al. 2003; Rasgon et al. 2006). For Lepidoptera, several studies have provided evidence for the effect of Wolbachia on variation and evolution of mtDNA. For example, a selective sweep has signif icantly reduced mtDNA diversity in Wolbachia infected Acraea encedana butterf lies (Jiggins 2003). Wolbachia was also found to inf luence the diversity of mtDNA in Polytremis nascens (Jiang et al. 2014). Because mtDNA diversity is widely used to infer the historical demography of insect populations, it is critical to know whether Wolbachia affect mtDNA variation of S. inferens or not.
In our study, measures of genetic variation of mtDNA in infected populations were not signif icantly lower or higher than that in the uninfected populations; no association was found between Wolbachia infection strains and phylogeny of mtDNA haplotypes; the mtDNA differentiation mainly existed within groups (divided as infected and uninfected) rather than among groups based on AMOVA analysis; no correlation between the genetic distances of hosts and symbionts was detected; mtDNA genes of S. inferens did not deviate signif icantly from neutral evolution according to nonsignif icant Tajima's D and Fu's F values. Consequently, our results reveal no signif icant effects of Wolbachia on host mtDNA evolution and diversity in natural S. inferens populations currently. One possible explanation is that Wolbachia only recently invaded this species and is in the process of spreading through the S. inferens population. This could also explain how uninfected and infected individuals existed in the same haplotype. It seems that the newly infected Wolbachia could not affect mtDNA evolution because it is well-known that the selective sweep may affect both uninfected and infected hosts (Yu et al. 2011), and whether this occurs will depend on the time that has elapsed since the symbiont invaded and on the transmission rate of the symbiont from mother to offspring (Turelli et al. 1992). Furthermore, it is possible that different strains possessed different functions or effects, and some specif ic strains could affect evolution of host while some couldn't. The fact is that in some host species, only particular Wolbachia strains infected the specif ic host. For example, in Drosophila melanogaster, Wolbachia occurs as the single strain w Mel, and the other Wolbachia genotypes are very rare and found only locally (Ilinsky 2013). Therefore, the Wolbachia strains in S. inferens might not currently have the ability to disrupt the evolution of mtDNA.
An interesting f inding of our study is the genetic differentiation of mtDNA related to the presence of different Wolbachia strains. Pairwise FSTvalues showed signif icant genetic differentiation between the uninfected and the infected group for w Pip and w Con strains as well as the three strains based on ftsZ gene. The reason could be that the low number of samples in each strain bias the signif icance.
5. Conclusion
Our f indings support that currently Wolbachia has no effects on the evolutionary dynamics of mtDNA variation in S. inferens. Therefore, mtDNA genes could be robust markers for population genetics research of S. inferens.
Acknowledgements
We sincerely thank our colleagues from Plant Protection and Quarantine Stations of Tangshan, Chongming, Xiangyang, Jingzhou, Ningxiang, Nanping, Yueqing, Guangyuan, Jiujiang, Dazhou, Yanhe, Guanyang, Yizhou, Zhaotong, Dehongzhou, Xishuangbanna, and Yunxiao, as well as Zhumadian Institute of Agricultural Science, Yuntai Farm in Lianyungang, Fuyang Normal University, Jiangxi Agricultural University, Sichuan Agricultural University, Guizhou Institute of Plant Protection, Yunnan Academy of Agricultural Sciences, Nanjing Agricultural University, Guangxi Academy of Agricultural Sciences, Sun Yat-Sen University, and Guangdong Ocean University for collecting Sesamia inferens samples.
We thank Dr. Roy Van Driesche from University of Massachusetts Amherst for polishing our manuscript. We are grateful to our colleague Dr. Carl E. Hjelmen from Texas A&M University for data analysis. The authors also would like to thank Testing Center of Yangzhou University, China for assistance. This work was supported by the National Key R&D Program of China (2017YFD0200400).
Appendicesassociated with this paper can be available on http://www.ChinaAgriSci.com/V2/En/appendix.htm
 Journal of Integrative Agriculture2019年5期
Journal of Integrative Agriculture2019年5期
- Journal of Integrative Agriculture的其它文章
- Characterization of TaCOMT genes associated with stem lignin content in common wheat and development of a gene-specif ic marker
- Phenotypic characterization and genetic mapping of the dwarf mutant m34 in maize
- Morphological diversity and correlation analysis of phenotypes and quality traits of proso millet (Panicum miliaceum L.) core collections
- Field identif ication of morphological and physiological traits in two special mutants with strong tolerance and high sensitivity to drought stress in upland rice (Oryza sativa L.)
- Crosstalk of cold and gibberellin effects on bolting and f lowering in f lowering Chinese cabbage
- Foliar spraying of aqueous garlic bulb extract stimulates growth and antioxidant enzyme activity in eggplant (Solanum melongena L.)