
毛细管电泳法测定硫酸多黏菌素B中的有关物质
2019-05-05 03:00张含智秦峰徐晓曦刘浩
中国抗生素杂志 2019年4期
张含智 秦峰 徐晓曦 刘浩
(上海市食品药品检验所,上海 201203)
多黏菌素B(polymyxin B sulfate,PMB,结构通式如图1所示)是一类由多黏芽孢杆菌发酵产生的多肽类抗生素混合物,其硫酸盐主要用于治疗多重耐药革兰阴性菌,被称为“抗击超级细菌的最后一道防线”[1-4]。PMB组分之间较为相似,其主要结构为N-端脂肪链(FA)、α,γ-二氨基丁酸及氨基酸(亮氨酸、异亮氨酸、苯丙氨酸、苏氨酸等)经酰胺键连接而成,4位及5位α,γ-二氨基丁酸结合形成环七肽,边链包括FA及三肽。已知的主要成分为PMB1、PMB2、PMB3及PMB1-I,差别在于FA取代基或氨基酸不同,具体区别见表1。

表1 多黏菌素B组分信息表Tab.1 The characteristics of known components in Polymyxin B
目前,中国药典(ChP)2015年版[5]、美国药典(USP)40版[6]、欧洲药典(EP)9.0版[7]均采用液相色谱法分析PMB中的组分及有关物质,通过相对保留时间对主要组分进行定性分析。对于PMB中的微量组分,Govaerts等[8-10]通过制备色谱及液质联用方法推断了约30种有关物质的结构。张含智等[11]根据HPLC-MS/MS推断了PMB中一种具有双键结构的组分,其结构需进一步鉴定。崔阿龙等[12-13]通过化学合成PMB中的单一组分,为构效关系研究及单一组分对照品制备奠定了基础,并揭示出某些单一组分的抗菌活性较总组分要高,且毒性要低。毛细管电泳(CE)方法对于复杂体系具有较好的分离效果,是液相色谱良好的正交补充方法。Kristensen等[14]将两性离子表面活性剂PAPS作为假固定相加入到缓冲液中,对PMB组分的分离选择性较好,但组分之间出峰时间较集中且主峰有明显的拖尾现象。Kang等[15]选择甲基-β-环糊精作为添加剂,建立了PMB的CE定性定量方法,该方法柱效高、稳健性好;本文采用该方法分析国产注射用硫酸PMB时,发现PMB1与相邻杂质的分离度较差。
为建立适用于国产硫酸PMB有关物质分析的CE方法,避免杂质对PMB定量产生影响,建立了羟丙基-β-环糊精(HP-β-CD)、异丙醇作为分离添加剂、三乙醇胺(TEA)-磷酸缓冲液的CE方法。通过优化HP-β-CD及异丙醇的含量,开展线性、检测限、精密度等方法学考察,确立了最优的分析方法,最终应用于硫酸PMB原料药及制剂有关物质的检测中,为国产样品的质量控制提供了一种可供借鉴的检测方法。
1 仪器与试药
Agilent G1600AX型高效毛细管电泳仪(美国Agilent Technologies公司),熔融石英毛细管(60cm,50μm,河北永年光导纤维厂)。
ChP 2015年版多黏菌素B(批号:130313-201310)标准品购自中国食品药品检定研究院(中检院,ZJY),USP硫酸多黏菌素对照品(批号:N1M425),硫酸多黏菌素B原料药(批号:A1411105)、注射用硫酸多黏菌素B(批号:1512801、1512802、1512803)均由上海第一生化药业有限公司提供。乙腈为色谱纯,三乙醇胺、磷酸和异丙醇均为分析纯,水为超纯水。α-CD、β-CD、甲基-β-CD、HP-β-CD均购自Sigma-Aldrich。
2 方法与结果
2.1 溶液配制
运行缓冲液:130mmol/L三乙醇胺溶液用磷酸调节pH值至2.5,加入5%(V:V)异丙醇与30mmol/L HP-β-CD,出口缓冲液瓶中不含HP-β-CD。
对照品溶液:精密称取硫酸PMB标准品(USP,PMB1含量69.7%)20mg,加2mL溶剂(水:乙腈=80:20)溶解,作为对照品储备液S1,浓度为10mg/mL,其中PMB1浓度为6.97mg/mL。
供试品溶液:精密称取硫酸PMB原料药及制剂50mg置于10mL量瓶中,加溶剂溶解,定容,摇匀,作为原料药供试品溶液,临用现制。制剂中含有50mg硫酸PMB,加溶剂溶解并全部转移至10mL量瓶中,定容,摇匀,作为制剂供试品溶液。
2.2 实验条件
工作电压24kV;柱温25℃;检测波长215nm;进样压力50mbar,进样时间3s。毛细管初次使用时,依次用1mol/L氢氧化钠溶液冲洗20min,蒸馏水冲洗5min,再用运行缓冲液冲洗20min。每次分析后用运行缓冲液冲洗3min。
在上述实验条件下,分别取对照品及供试品溶液进样分析。
2.3 线性及精密度试验
取对照品储备液S1经溶剂逐级稀释至PMB1的浓度为3.485(S2)、1.394(S3)、0.697(S4)、0.3485(S5)、0.0697(S6)、0.03485(S7)和0.01394mg/mL(S8),分别进样测定并记录电泳图,以PMB1峰面积A对浓度c(S6-S1)进行线性回归,得回归方程为:A=103.2c+4.3062,R2=0.9999。
精密度试验中,PMB1峰面积及迁移时间的天内重复性的RSD值分别为0.53%和0.60%(n=6),天间的RSD值分别为0.65%和0.69%(n=3),说明本方法的重复性较好。LOD(S/N=3)为13.94μg/mL,LOQ(S/N=10)为34.85μg/mL。
2.4 样品测定
取中检院标准品、USP对照品、企业原料药及3批制剂进行测定,并对结果进行比较分析。
3 结果与讨论
由于PMB属于发酵产品,不同的菌种及生产环境导致组分之间的差异较大,已有方法可能不适用于不同企业的PMB的分析。如采用文献[14]中的CE方法检测上海第一生化药业有限公司的硫酸PMB时,组分出峰时间较为集中,且B1与相邻峰的分离并不理想,见图2A;采用文献[15]的方法,B1、B1-I分别与相邻杂质未实现基线分离,改变其中的三乙醇胺、甲基-β-CD及异丙醇的含量,未有明显的改善效果,见图2B,且两种方法均存在主峰拖尾的情况。本研究在此基础上,通过筛选其他不同的环糊精(如α-CD、β-CD、HP-β-CD),发现HP-β-CD较适合于PMB各组分的分离,并考察了三乙醇胺、HP-β-CD及异丙醇对分离的影响,筛选出最优的缓冲液条件。
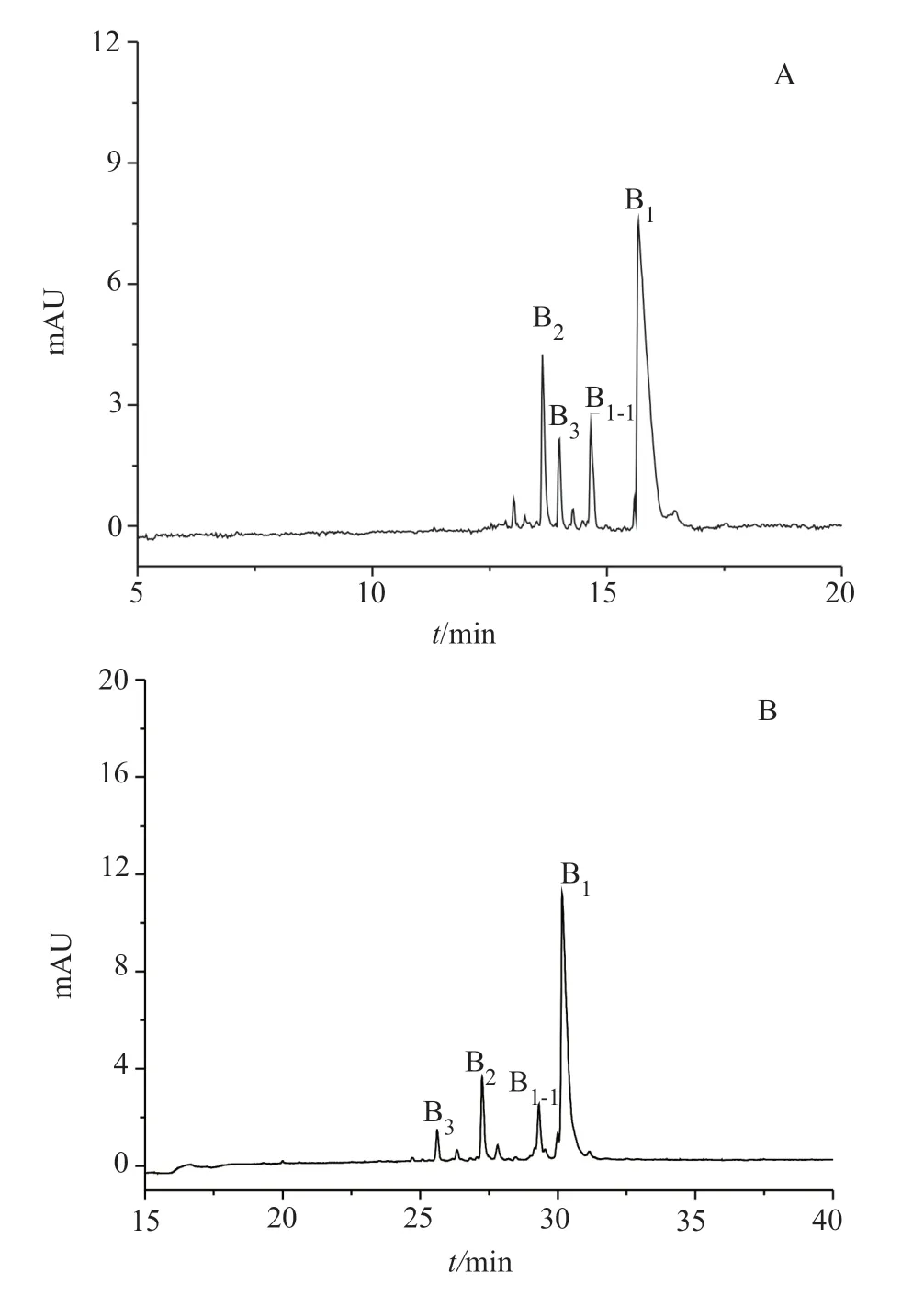
图2 PMB分离的典型色谱图Fig.2 The typical chromatogram of PMB
3.1 缓冲液组成对分离效果的影响
三乙醇胺缓冲液经H3PO4调节pH至2.5后,熔融石英毛细管管壁的硅羟基在该条件下几乎不解离,避免了电渗流对分离的影响。PMB各组分根据各自不同的电泳淌度进行分离,同时由于降低了正电性多肽在管壁的吸附,提高分析重复性。
固定三乙醇胺(130mmol/L)及HP-β-CD(30mmol/L)的含量,考察缓冲液中异丙醇的含量对分离选择性的影响。缓冲液中无异丙醇时,PMB2、Ile-PMB1与PMB1在较集中的时间内迁移出峰,掩盖了较多的微量杂质。随着异丙醇含量的增加,PMB各组分之间的分离度增大,说明该有机溶剂对分离有明显地改善效果;但分析时间延长,如异丙醇为10%时,PMB1迁移时间为38min。最终确定缓冲液中异丙醇的含量为5%。
当固定三乙醇胺(130mmol/L)及异丙醇(5%)的含量时,发现HP-β-CD的含量对PMB的分离选择性具有明显的影响。调整HP-β-CD含量从10、20和30mmol/L变为40mmol/L,随着HP-β-CD浓度的增加,PMB各组分的迁移时间逐渐增大,见图3,说明PMB与HP-β-CD之间的相互作用增强,导致组分之间的分离选择性增大。PMB1与PMB1-I逐渐分离开,两者之间的微量杂质峰被检测到;但PMB2与PMB1-I之间的分析时间缩短,导致HP-β-CD在40mmol/L时,有较多的微量杂质被包埋在PMB1-I峰中。综合考虑以上因素,确定HP-β-CD浓度为30mmol/L。
3.2 CE对不同硫酸PMB的检测结果
采用“3.1”项中筛选到的最优的分离条件,即缓冲液中含有30mmol/L HP-β-CD、5%异丙醇的130mmol/L三乙醇胺溶液(用磷酸调节pH至2.5),对中检院标准品、USP对照品、硫酸PMB原料药及制剂进行检测。按照ChP 2015年版中硫酸多黏菌素B有关物质项下规定,单个杂质不得过3.0%,杂质总量不得过17.0%,根据峰面积归一化计算以上样品中单个杂质及杂质总量,见表2。经对比发现,企业样品中的单个杂质及杂质总量均在规定限度之内。与液相方法(按照ChP 2015年版硫酸多黏菌素B有关物质项下的分析方法)结果进行比较,最大单个未知杂质之间的差别较小,但总杂质含量差别较大,可能原因是在毛细管电泳中进样体积较液相进样体积小,造成某些微量杂质未被检测到,应需要增加进样体积。
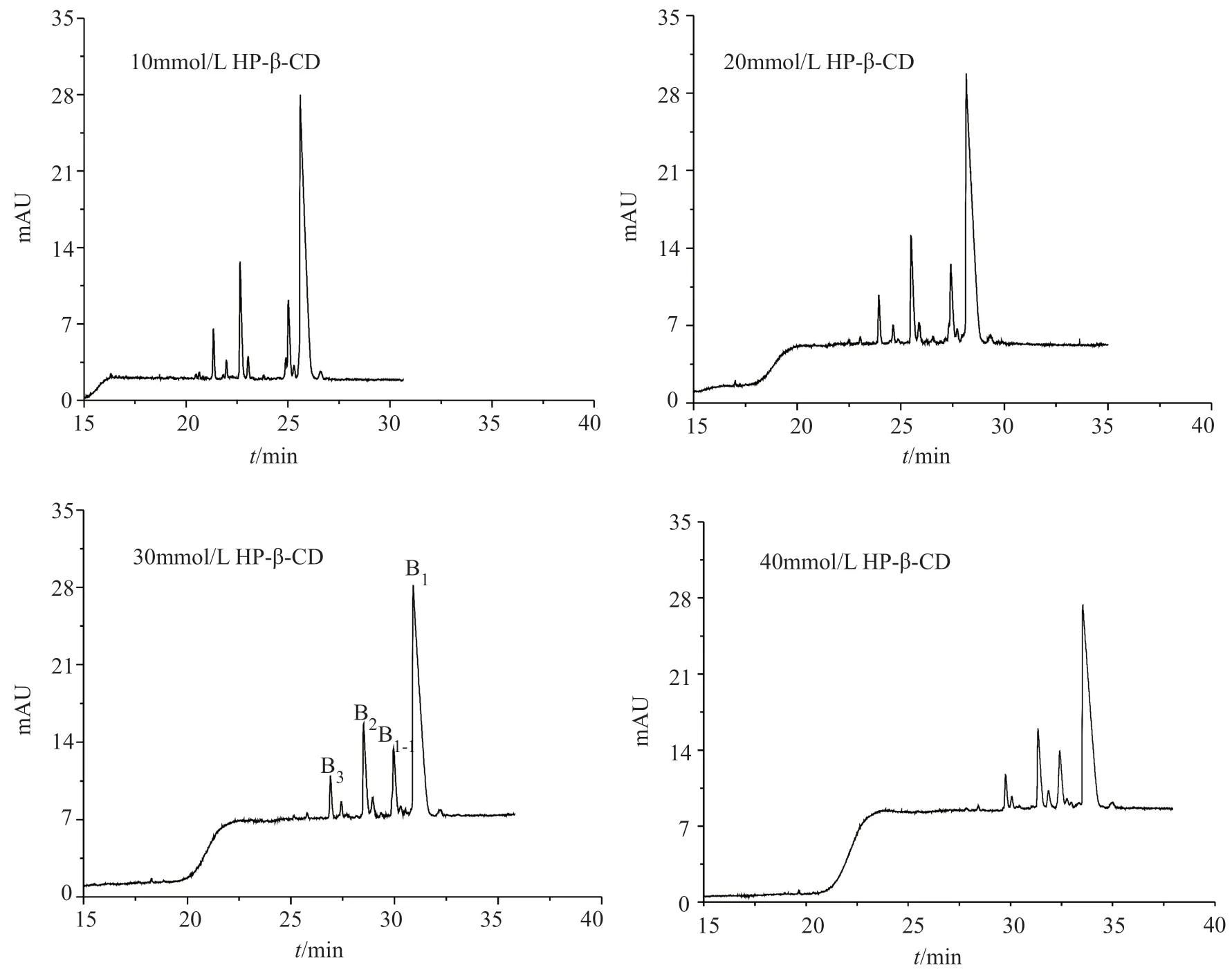
图3 HP-β-CD对PMB分离选择性的影响Fig.3 The effect of HP-β-CD on the separation selectivity of PMB
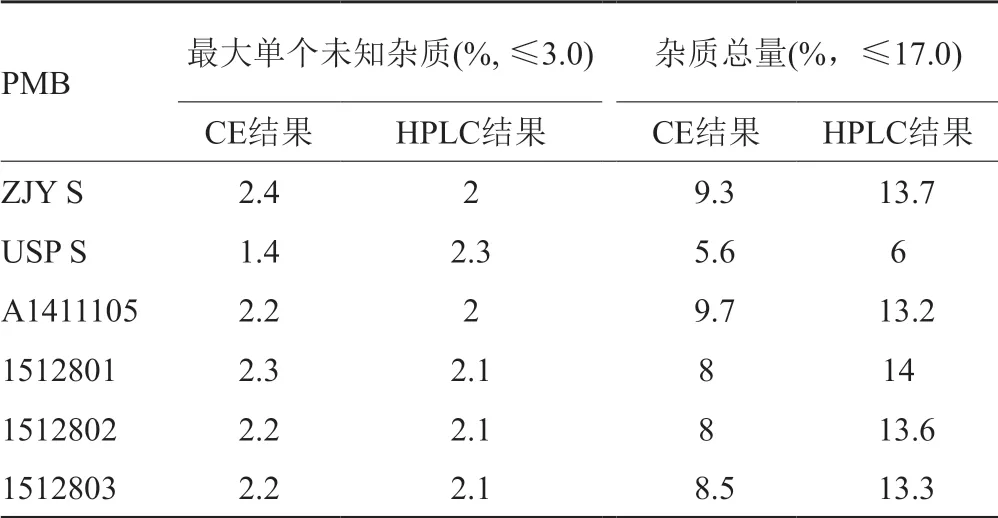
表2 PMB中有关物质的含量Tab.2 The contents of relative substances in PMB
4 结论
本研究开发了一种CE方法用于PMB中的有关物质分析,采用HP-β-CD作为缓冲液改性剂可以提高分离的选择性。本法灵敏度较高、重复性较好,可以作为药典中液相方法的补充方法对PMB中的有关物质进行定量分析,为国产药品的质量控制提供了一种可供选择的方法。
猜你喜欢
可再生能源(2021年12期)2021-12-28
农业与技术(2021年21期)2021-11-17
天津医科大学学报(2021年4期)2021-08-21
日用电器(2019年7期)2019-08-07
智能制造(2019年10期)2019-03-23
中国感染与化疗杂志(2019年4期)2019-01-06
环球时报(2018-02-02)2018-02-02
建材发展导向(2016年6期)2017-01-17
科学与财富(2016年28期)2016-10-14
印刷技术·数字印艺(2015年10期)2015-12-10
