
朝鲜蓟中2种木犀草素类化合物液相制备条件优化
2019-04-29 03:31师明月曹清明李群钟文惠王元清刘志文张喜平
食品与机械 2019年3期
师明月曹清明李 群钟文惠王元清刘志文张喜平
(1. 中南林业科技大学食品科学与工程学院,湖南 长沙 410004;2. 汇美农业科技有限公司, 湖南 常德 415137;3. 常德市农林科学研究院,湖南 常德 415000)
朝鲜蓟是一种高营养价值的保健蔬菜[1-2],被誉为“蔬菜之皇”[3-4],其提取物一直用于民间医药[5],在各种药理试验中表现出促进消化[6]、保肝[7]、利胆[8]、抗癌[9]以及抑制低密度脂蛋白氧化[10]的能力。临床试验表明,朝鲜蓟具有降低血浆胆固醇[11-12]、降低肠易激综合征[13]、减肥[14]以及控制空腹血糖升高[15]等功效。
朝鲜蓟的次生代谢产物主要是多酚类化合物[16-18]和具有苦味的愈创木烷倍半萜内酯类化合物。多酚类化合物表现出抗氧化能力等活性,愈创木烷倍半萜内酯表现出抗炎及抑制肿瘤等多种功效[19-20]。
Pandino等[21]报道了6个品种朝鲜蓟的3个部位(包括花托、外苞片和内苞片)的多酚含量,研究结果表明木犀草素糖苷类化合物含量为24(Tema 2000品种的外苞片)~620 mg/kg(Tondo di Paestum品种的花托),其含量比咖啡酰奎宁酸类化合物含量[23(Tema 2000品种的内苞片)~5 771 mg/kg(Violetto di Sicilia品种的内苞片)]和芹菜素含量[870(Blanc Hyerois品种的内苞片)~5 397 mg/kg(Tema 2000品种的花托)]低。木犀草素(luteolin)类化合物含量虽低于其他2类酚类化合物,但由于其B环具有邻二酚羟基结构,表现出强抗氧化活性、抑制胆固醇合成、抑制糖尿病以及抗肿瘤活性等功效。Schlupper等[22]研究木犀草素及其衍生物的活性发现,木犀草素、木犀草素7-O-糖苷在极低浓度下表现出明显的抗氧化能力;Gebhardt[23]研究表明木犀草素类化合物的木犀草素配苷主要负责抑制肝脏胆固醇的生物合成,而绿原酸、咖啡酸、洋蓟素和其他二咖啡酰奎宁酸无显著影响;且木犀草素亦能有效阻断胰岛素对胆固醇生物合成的影响,从而验证了朝鲜蓟的降血脂作用。木犀草素对表皮生长因子受体—酪氨酸激酶(EGFR-TK)具有明显抑制作用。Rahimuddin等[24]研究发现木犀草素及其糖苷对UVA导致的皮肤成纤维细胞脂质过氧化具有抑制作用。
目前,中国种植的朝鲜蓟品种主要有:改良绿球、绿宝石、帝王之星、A106、A109、洛尔卡和上海朝鲜蓟等[25]。洛尔卡(Lorca)是常德市于2005年从法国引进的一个加工型朝鲜蓟新品种,并通过常德市农业科学研究所、汇美农业科技有限公司选育,经过多年试验与示范,目前已成为湖南省的主要栽培品种[26]。课题组拟以洛尔卡朝鲜蓟作为研究对象,开发木犀草素类化合物的分离纯化新方法。
1 材料与方法
1.1 材料与试剂
朝鲜蓟(CynarascolymusL.)花苞:洛尔卡(Lorca)品种,常德汇美农业科技有限公司提供。
大孔树脂:AB-8型,安徽三星树脂科技有限公司;
木犀草苷-7-β-D-芸香糖苷标准品:HPLC纯,纯度≥98.0%,美国Sigma-Aldrich公司;
木犀草素7-O-β-D-葡萄糖苷标准品:HPLC纯,纯度≥98.0%,美国Sigma-Aldrich公司;
氘代试剂CH3OD:氘代率≥99.8%,美国Sigma-Aldrich公司。
1.2 仪器与设备
RP-C18层析柱:Sepax GP-C18型,粒径40~60 μm,苏州赛分科技有限公司;
Sepax GP-C18柱:4.6 mm×150 mm,40~60 μm,自制;
Venusil MP C18柱:4.6 mm×250 mm,5 μm,天津博纳艾杰尔科技有限公司;
Venusil MP C18柱:10 mm×250 mm,5 μm,天津博纳艾杰尔科技有限公司;
Unitary C18柱:4.6 mm×250 mm,5 μm,华谱新创科技有限公司;
高效液相色谱仪:LC3000型(配备Newstyle NU3000 Serials UV/VIS检测器),北京创新恒通科技有限公司;
高效液相色谱:2695-2996型,美国Waters公司;
质谱仪:UHPLC-QTOF/1290-6530型,美国安捷伦科技有限公司;
核磁共振仪:Bruker AVANCE-500MHz型,德国Bruker公司。
1.3 方法
1.3.1 朝鲜蓟粗提液的制备 将朝鲜蓟晒干,粉碎,得到1.1 kg朝鲜蓟粉末。在室温下,将1.1 kg朝鲜蓟粉末按料液比1∶10(g/mL)的比例,用70%乙醇浸提72 h。过滤,得到2 L粗提液。
1.3.2 液相色谱条件 对朝鲜蓟粗提液进行液相条件摸索:进样量20 μL,流动相为甲醇-0.1%甲酸水,流速1 mL/min,检测波长205 nm时,峰的数量较多,为朝鲜蓟提取液分析的较好条件,如无另外说明,将采纳该条件。
1.3.3 AB-8型大孔树脂对提取物进行粗分 采用大孔树脂吸附法(AB-8型)对朝鲜蓟粗提液(1 L)进行分离纯化。具体步骤:将1 L柱体积的大孔树脂颗粒进行预处理后装柱,用水冲至无醇味,上样过程中,接收流出液进行液相色谱检测(分析上样量是否超载),洗脱梯度为0~30 min,流动相A由5%增至100%。待样品完全吸附后,依次用水,20%,50%,80%乙醇进行洗脱,洗脱至无色或颜色较浅,将接收的流出液依次编号为:A1、A2、A3……,经高效液相色谱检测进行合并,洗脱梯度为0~30 min,流动相A由5%增至100%。
1.3.4 RP-C18对A27~29组分进行细分 用Sepax GP-C18填料(40~60 μm)自制液相柱优化洗脱程序,以确定RP-C18层析柱的洗脱程序。将4 g冷冻干燥样品(A27~29组分)用50%甲醇完全溶解,浓度为40 mg/mL。根据分离度选择优化洗脱条件。将液相优化程序用于柱层析洗脱程序。将接收的流出液依次编号为:C1、C2、C3……。
1.3.5 组分C16~19的液相半制备 由于半制备时液相色谱采用的色谱柱的灵敏度和分离度都较低,故在进行液相半制备之前,需对半制备条件进行摸索。试验以分离度和峰型为优化目的,设置了5种流动相组成(体积比),流动相1、流动相2、流动相3、流动相4和流动相5分别为60∶40的甲醇-0.1%甲酸水、45∶55的甲醇-0.1% 甲酸水、40∶60的乙腈-0.1%甲酸水、30∶70的乙腈-0.1%甲酸水、23∶77的乙腈-0.1%甲酸水,比较分析以获得最佳流动相分析条件。待条件优化后,采用Venusil MP C18(10 mm×250 mm,5 μm)半制备柱,流速增至3 mL/min进行放大制备。为避免样品浪费,缩短制备时间,还需确定进样量,试验设置了30,50 μL进样量进行研究。
1.3.6 质谱条件
(1) 质谱工作站软件:Agilent MassHunter B.06.00版。色谱工作站软件:Agilent 1200化学工作站。
(2) LC-MS工作条件:Unitary C18(4.6 mm×250 mm,5 μm)色谱柱,洗脱剂乙腈—水(体积比25∶75),进样量2 μL,检测波长254 nm;电喷雾离子源,正、负离子检测。
1.3.7 核磁共振检测条件 溶剂为CH3OD,NMR的工作频率分别为1H谱500 MHz,13C谱为125 MHz。1H-NMR 谱采样脉冲宽度90°,累加32次;13C-NMR谱采样脉冲宽度90°,累加2 000次。
2 结果与分析
2.1 AB-8型大孔树脂对样品进行粗分
根据HPLC图的峰型和出峰时间进行合并,A27~29组分出峰相对简单(如图 1所示),3个组分出峰相似,合并,浓缩,冷冻干燥,得到4 g目标组分A27~29。
2.2 RP-C18对A27~29组分进行细分
优化的洗脱条件见表1,此条件下分离效果较好。将此条件用于A27~29组分的洗脱,即:依次用30%,48%,100%甲醇进行洗脱。通过计算,30%甲醇用量为6 L,48%甲醇用量为4 L,100%甲醇用量为2 L。根据液相检测结果合并为C1、C2~4、C5~10、C11~15、C16~19……,分别减压浓缩,冷冻干燥,得到C16~19组分0.32 g。图2中,组分C16~19出峰较为简单,能够分离出纯化合物,考虑对C16~19组分进行液相半制备。
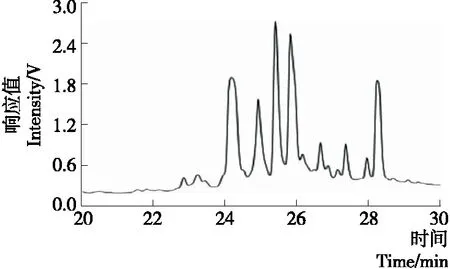
图1 A29组分的HPLC分析图

表1 HPLC洗脱程序

图2 C16~19组分的HPLC分析图
2.3 组分C16~19的液相半制备
2.3.1 液相条件的优化
(1) 流动相的优化:对5个不同流动相进行比较,得到乙腈∶0.1%甲酸水(体积比)=23∶77(流动相5)为最优流动相。
由图3(a)可知,在流动相1条件下,甲醇浓度为60%,由于甲醇浓度过高,造成各峰一起被洗脱,且未达到基线分离,故需要降低甲醇浓度。图3(b)中,在流动相2条件下,降低甲醇浓度,分离效果仍不太理想,故改用乙腈作为流动相,40%乙腈浓度(流动相3)色谱图如3(c),峰一下被洗脱,需降低乙腈浓度,故改用流动相4(30%乙腈),出峰如图3(d),各峰集中,较快被洗脱,说明30%乙腈浓度过高,将乙腈浓度降低到23%(流动相5),如图3(e)所示,各峰的分离效果比较好。故优化的液相分析条件:流动相A为乙腈(23%),B为0.1%甲酸水(77%);优化的洗脱条件见表2。
(2) 分离度比较:为判断物质在色谱柱中的分离情况,常用分离度作为柱的总分离效能指标,分离度越大则表明物质在色谱柱中的分离效果越好。对上述的5种不同的流动相分析条件所得的图谱进行分离度比较(以色谱图中第一、第二2个峰之间的分离度计算),结果如表3所示。
从图2(e)、表3可知,在乙腈∶0.1%甲酸水为23 ∶77(体积比)的条件下,分离度较大,分离效果最好,且峰形尖锐,保留时间合适,对环境有害的有机相使用比例更少。
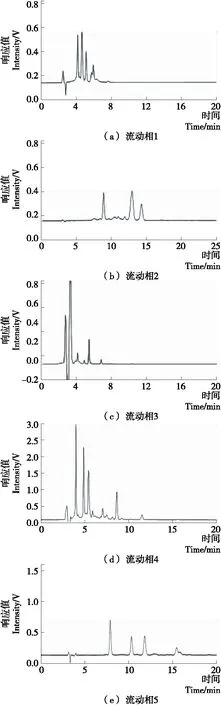
图3 C16~19组分在5种不同流动相下的HPLC分析图

表2 优化的C16~19组分HPLC洗脱程序
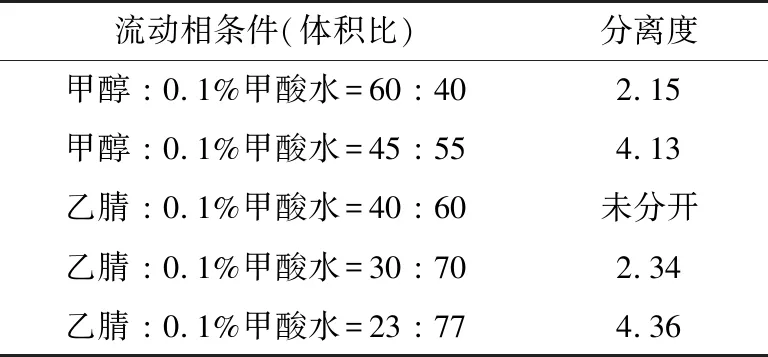
表3 5种分离条件下的分离度
(3) 制备条件的确定:利用上述优化的条件,除将柱子改为半制备柱[Venusil MP C18(10 mm×250 mm,5 μm),流速改为3 mL/min]外,其他条件不变,即洗脱程序为表2所示。将C16~19组分用23%乙腈溶解,分别对进样量30,50 μL进行优化,所得制备图(50 μL进样量色谱见图4)差异较小,为节省溶剂,缩短试验时间及损耗,本试验选择进样量为50 μL。
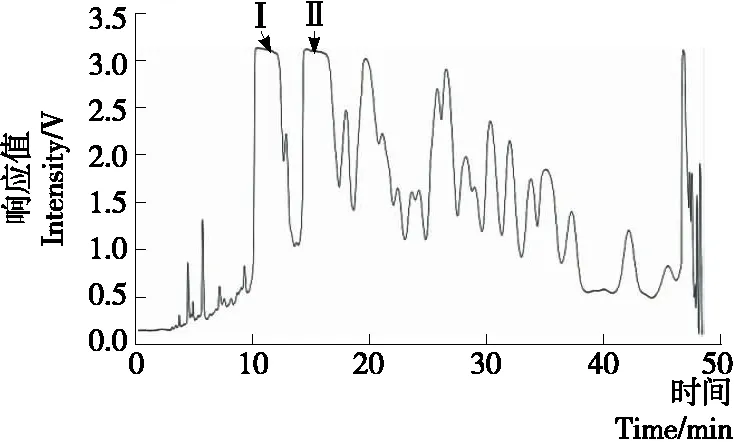
图4 C16~19组分的HPLC半制备图(50 μL)
综上所述,半制备条件为:流速3 mL/min,进样量50 μL,流动相A为乙腈,流动相B为0.1%甲酸水,洗脱程序如表2所示(40 min后的梯度为冲洗色谱柱)。
2.3.2 液相半制备 将0.32 g C16~19组分用23%乙腈溶解,浓度为200 mg/mL,按优化的条件制备。如图4所示,峰I、II分离效果较好,分别收集峰I、II。将收集液减压浓缩,冷冻干燥后,称重,液相色谱测定纯度,并得到纯度较高的2个化合物,记为H-01、H-02,其质量分别为30,15 mg,提取得率分别为54.5,27.3 mg/kg,纯度分别为97.8%,96.5%。2个样品纯度均符合核磁检测条件。
2.4 化合物的结构解析
2.4.1 H-01 ESI-MS给出:正离子模式下,质谱数据m/z595.165 7[M+H]+;负离子模式下,质谱数据m/z593.149 2[M-H]-,推测其分子式为C27H30O15。1H-NMR:6.52(1H,s,H-3), 6.46(1H,d,J=2 Hz,H-6),6.63(1H,d,J=2 Hz,H-8),7.33(2H,m,H-2′,6′),6.88(1H,s,H-5′),5.00(1H,m,H-1″),3.64(1H,m,H-6″a),4.05(1H,d,J=9.7 Hz,H-6″b),4.73(1H,s,H-1‴),3.93(1H,s,H-2‴),3.76(1H,d,J=9.4 Hz,H-3‴),1.20(3H,d,J=5.8 Hz,H-6‴)。13C-NMR:166.73(C-2), 104.14(C-3),183.85(C-4),162.76(C-5),101.07(C-6),164.57(C-7),96.16(C-8), 158.67(C-9),107.00(C-10),123.37(C-1′),116.85(C-2′),146.84(C-3′),151.06(C-4′),114.29(C-5′),120.62(C-6′),102.05(C-1″),74.71(C-2″),77.73(C-3″),71.31(C-4″),77.09(C-5″),67.47(C-6″),101.54(C-1‴),72.37(C-2‴),72.03(C-3‴),74.03(C-4‴),69.77(C-5‴),17.91(C-6‴)。
综合所有核磁谱图及质谱结果,H-01鉴定为Luteolin 7-O-α-L-Rhamnosyl(1-6)-β-D-Glucoside(木犀草素7-O-α-L-鼠李糖(1-6)-β-D-葡萄糖苷),将其1H-NMR、13C-NMR以及质谱数据与文献[27]报道的化合物3对照,基本一致。
2.4.2 H-02 质谱数据m/z449.109 4[M+H]+、碎片离子287.057 5+,推测其分子式为C21H20O11,含有一个黄酮骨架和一个葡萄糖糖苷。1H-NMR:6.56(1H,s,H-3),6.45(1H,s,H-6),6.74(1H,s,H-8),7.37(1H,s,H-2′),6.89(1H,d,J=8.0 Hz,H-5′),7.38(1H,d,J=8.0 Hz,H-6′),5.07(1H,d,J=7.0 Hz,H-1″), 3.50~3.60(2H,m,H-2″,H-3″),3.40(1H,m,H-5″),3.96(1H,d,J=12.2 Hz,H-6″a),3.72-3.75 (1H,dd,J=12.2,5.6 Hz,H-6″b)。13C-NMR:166.82(C-2),107.08(C-3),183.99(C-4),164.74(C-5),101.16(C-6),,162.82(C-7),96.07(C-8),158.93(C-9),104.18(C-10),123.48(C-1′),114.31(C-2′),147.03(C-3′),151.16(C-4′),116.80(C-5′),120.53(C-6′),101.64(C-1″),74.75(C-2″),77.86(C-3″),71.29(C-4″),78.39(C-5″),62.49(C-6″)。
综合所有核磁谱图及质谱结果,H-02鉴定为Luteolin 7-O-β-D-Glucoside(木犀草素7-O-β-D-葡萄糖苷),将其1H-NMR、13C-NMR及质谱数据与文献[28]报道的化合物1对照,基本一致。
2.5 C16~19组分中木犀草素含量的确定
采用表2优化的液相条件,将木犀草苷-7-芸香糖苷和木犀草素7-O-β-D-葡萄糖苷标准样品分别用乙腈溶解,进行液相分析。采用外标法,对C16~19组分中的木犀草苷-7-芸香糖苷和木犀草素7-O-β-D-葡萄糖苷进行定量,结果表明,2个化合物在C16~19流分中的含量分别为10.4%,5.1%,C16~19流分中H-01和H-02制备的得率分别为90.1%,91.9%,说明该液相制备方法的优化合理,可应用于类似组分的分离制备。
3 结论
本研究以洛尔卡(Lorca)为研究对象,开发了木犀草素类化合物的分离纯化新方法,得到纯度分别为97.8%和96.5%的木犀草苷-7-芸香糖苷和木犀草素7-O-β-D-葡萄糖苷,其总提取得率分别为54.5,27.3 mg/kg,与Pandino等[21]研究的朝鲜蓟相比,本试验中所采集的样品木犀草素类化合物含量中等,若能选择木犀草素类化合物含量高的样品,可制备更多的纯品。木犀草素糖苷由于B环具有3’,4’邻二酚羟基结构,其具有较强的抗氧化功能,分离和纯化木犀草素糖苷具有非常重要的意义。
猜你喜欢
煤化工(2022年3期)2022-07-08
色谱(2021年7期)2021-06-07
天然产物研究与开发(2018年2期)2018-04-04
中成药(2017年12期)2018-01-19
西江月(2017年4期)2017-11-22
小雪花·成长指南(2016年10期)2016-11-01
山东工业技术(2016年10期)2016-09-06
中国资源综合利用(2016年10期)2016-01-22
中国烟草学报(2012年3期)2012-04-10
郑州大学学报(理学版)(2012年4期)2012-03-25
