
Mg-F受主-施主共掺SnO2的第一性原理研究
2019-04-28 08:53何海英张召君陈雅丽吴梓彬李杰森
原子与分子物理学报 2019年5期
何海英, 张召君, 陈雅丽, 吴梓彬, 李杰森
(1. 佛山科学技术学院 材料科学与能源工程学院, 佛山 528000; 2. 中山大学 材料学院, 广州 510275; 3.佛山科学技术学院 环境与化学工程学院 佛山 528000)
1 引 言
近年来,透明金属氧化物作为新一代半导体材料备受关注,其代表材料如氧化锌(ZnO)、氧化铟(In2O3)、氧化锡(SnO2)等. 然而,In是稀缺资源,成本高昂,研究可替换材料尤为重要;ZnO系与SnO2系是较好的候选材料,但掺杂ZnO自补偿效应严重;SnO2具有带隙宽(3.6 eV),可见光区透过率高,激子束缚能大(130 meV),化学稳定性良好等优点,被广泛应用于液晶显示、太阳能电池、发光二极管等众多光电器件领域. 一般情况下,因受到本征缺陷的影响,如氧空位(VO)和锡填隙(Sni),SnO2主要表现为n型导电类型[1],高价元素(Sb5+)替换Sn[2-4]或低价元素(F-)替换O[5-7],施主杂质掺杂引入电子载流子形成光电性能良好的n型导电SnO2. 然而,单极性限制了SnO2在半导体领域的实际应用,性能相当的p型材料成为研究热点. 故而,实现掺杂SnO2的p型转变是研究SnO2基光电器件的关键技术. Van de Walle研究小组通过计算预测IIIA族元素(Al、In、Ga)可实现SnO2的受主掺杂[8, 9]. 研究者们也分别从实验上制备了p型SnO2:Al[10, 11]、SnO2:Ga[12, 13]和SnO2:In[14, 15]. P型SnO2薄膜受主杂质掺杂浓度高达1021-2023cm-3,但是空穴浓度仅在1015-1019数量级[10-15]. 一方面空穴载流子被本征缺陷俘获,另外一方面杂质除了替换SnO2晶格中的Sn成为受主掺杂外,部分杂质进入SnO2晶格处于填隙状态充当施主杂质[16-18]. 要实现性能良好的p型SnO2掺杂,降低受主掺杂的形成能并形成稳定掺杂结构尤为重要,单受主掺杂已不能满足其要求. Yamamoto[19]提出同时掺入一定量的p型和 n型掺杂剂可以降低形成能,共掺杂可以有效地提高掺杂剂的溶解度. 相对于单受主掺杂,共掺杂还能有效改善SnO2的性质[20-22],Nb-Zr共掺杂SnO2体系具有磁性,Sr-F共掺杂SnO2具有良好的导电性,Fe-S共掺杂使SnO2呈现半金属性质. 课题组在前期研究中制备了p型Mg掺杂SnO2[23],为了使Mg形成更加稳定的受主掺杂,本文基于密度泛函的平面波超软赝势法研究Mg,F共掺情况下SnO2的电子结构及性质.
2 模型构建与计算方法
本文计算模型为SnO2金红石结构,四方晶系,晶格常数为a=b=4.737 Å,c=3.186 Å,空间群为P42/mnm,其晶体结构如图1(a)所示,其中,每个Sn原子位于6个O原子构成的八面体中心. 本文采用2×2×3的SnO2超胞结构进行相关掺杂计算研究,利用Mg原子替换Sn原子(MgSn)形成单受主掺杂,如图1(b)所示;利用F替换距离Mg原子最近的O原子(MgSn-FO)形成受主-施主共掺杂,如图1(c)所示. Mg和F的掺杂浓度分别是4.17%、2.08%.
本文采用Materials Studio 软件的CASTEP模块[21]进行基于密度泛函(DFT)的第一性原理计算. 经收敛性测试,截断能参数设置为400 eV,布里渊区k点设置为3×3×3. 研究的主要元素的电子组态为:O 2s22p4,Sn 5s25p2,Mg 3s2,F 2s22p5.
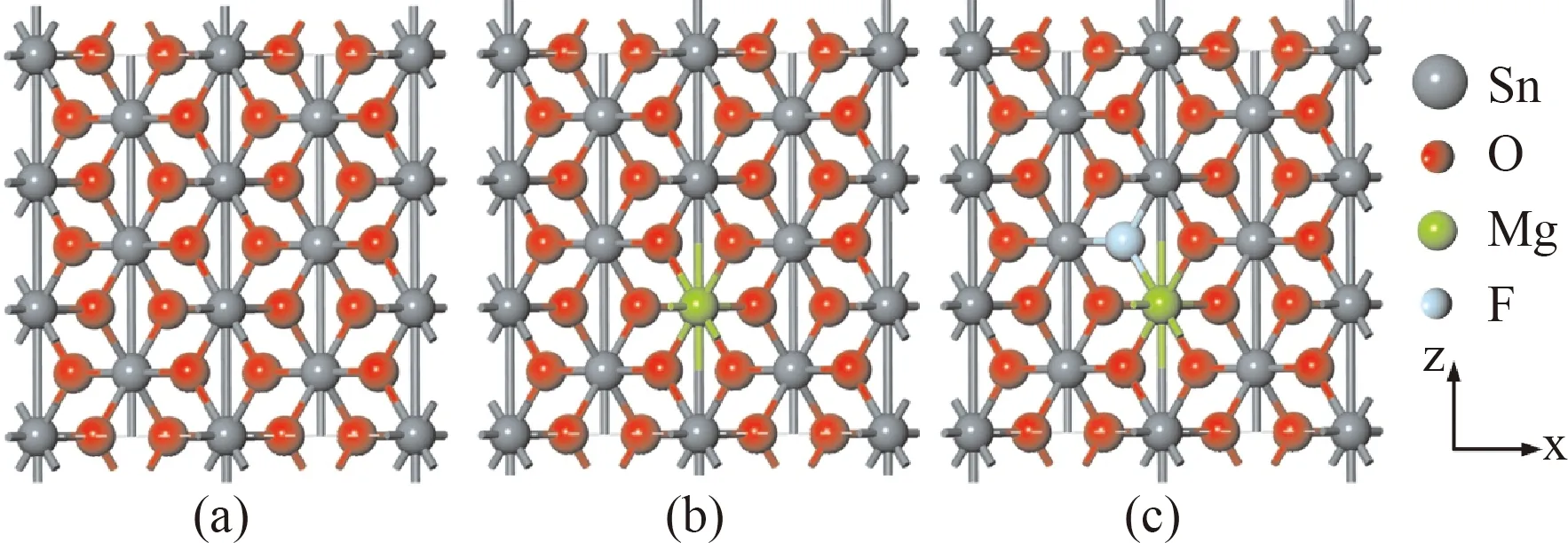
图1 SnO2超胞(a)不掺杂;(b)Mg掺杂;(c)Mg-F共掺杂Fig. 1 Supercell of SnO2 (a) un-doped, (b) Mg-doped, (c) Mg-F co-doped
3 结果与讨论
3.1 结构的稳定性
图2所示为SnO2及其掺杂模型局部结构. SnO2晶体中每个Sn原子被6个O原子包围形成八面体结构,O原子处于两种不同的位置,O1位于赤道平面,O2位于轴向,故而存在两种不同的Sn-O键,其中Sn-O1键键长为2.117 Å,Sn-O2键键长为2.139 Å. 掺杂杂质引入后导致了局部晶格畸变,Mg单受主掺杂时,与Mg临近的Sn-O键键长均略微缩短. 在受主掺杂的基础上进一步在临近Mg原子附近引入杂质F形成共掺杂体系时,由于多种杂质的引入不可避免的在一定程度上增大了SnO2晶体的局部晶格畸变,如图2(c)所示. 为了更加了解掺杂对SnO2晶体结构的影响,我们计算了掺杂前后SnO2的晶格参数,如表1所示. 计算体系采用的SnO2晶体的晶格参数是a=4.737 Å,c=3.186 Å[24],为了计算结构的准确性,必须首先进行结构优化,后续计算基于结构优化后的晶体结构进行. 结构优化后晶格参数略有变大,这主要是由于广义梯度近似(GGA)的固有因素导致[25]. 与优化后的晶格常数相比较,单受主Mg掺杂后晶格参数的变化率约为~1%,Mg-F共掺杂时晶格参数的变化率也约为~1%. 掺杂后局部的晶格畸变未能影响整个SnO2晶体晶格参数较大的变化,Mg-F掺杂能有一定的稳定性.
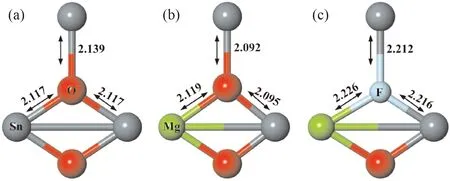
图2 SnO2及其掺杂模型局部结构(a)不掺杂;(b)Mg掺杂;(c)Mg-F共掺杂Fig. 2 Local structure of SnO2 (a) un-doped, (b) Mg-doped, (c) Mg-F co-doped
3.2 电子结构
图3是未掺杂、Mg掺杂和Mg-F共掺杂情况下SnO2靠近价带顶和导带底的能带结构图,费米能级位于0 eV. 由能带图得知,SnO2属于直接带隙半导体,未掺杂时直接计算出本征SnO2的带隙宽度为1.035 eV,低于实验值3.6 eV,然而与其他采用此方法计算的结构相近[26],广义梯度近似在计算过程中会低估带隙宽度,但GGA方法除了低估体系的禁带宽度外,但在同一个计算体系中,当计算环境相同时,不影响对变化趋势的分析,计算出的禁带宽度的值之间可以比较,因此本文中继续采用该方法计算. 对比未掺杂本征态和掺杂态,随着Mg的掺杂,增加了禁带宽度,进一步验证了实验测得的结果[23]. Mg-F共掺杂时,导带下移,F的引入使得禁带窄化,但费米能级并没有进入导带,掺杂体系仍然保持为p型导电类型,如图3(c)所示.
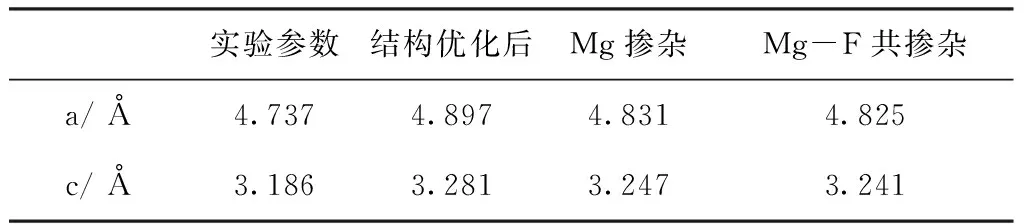
表1 SnO2掺杂前后的晶格常数
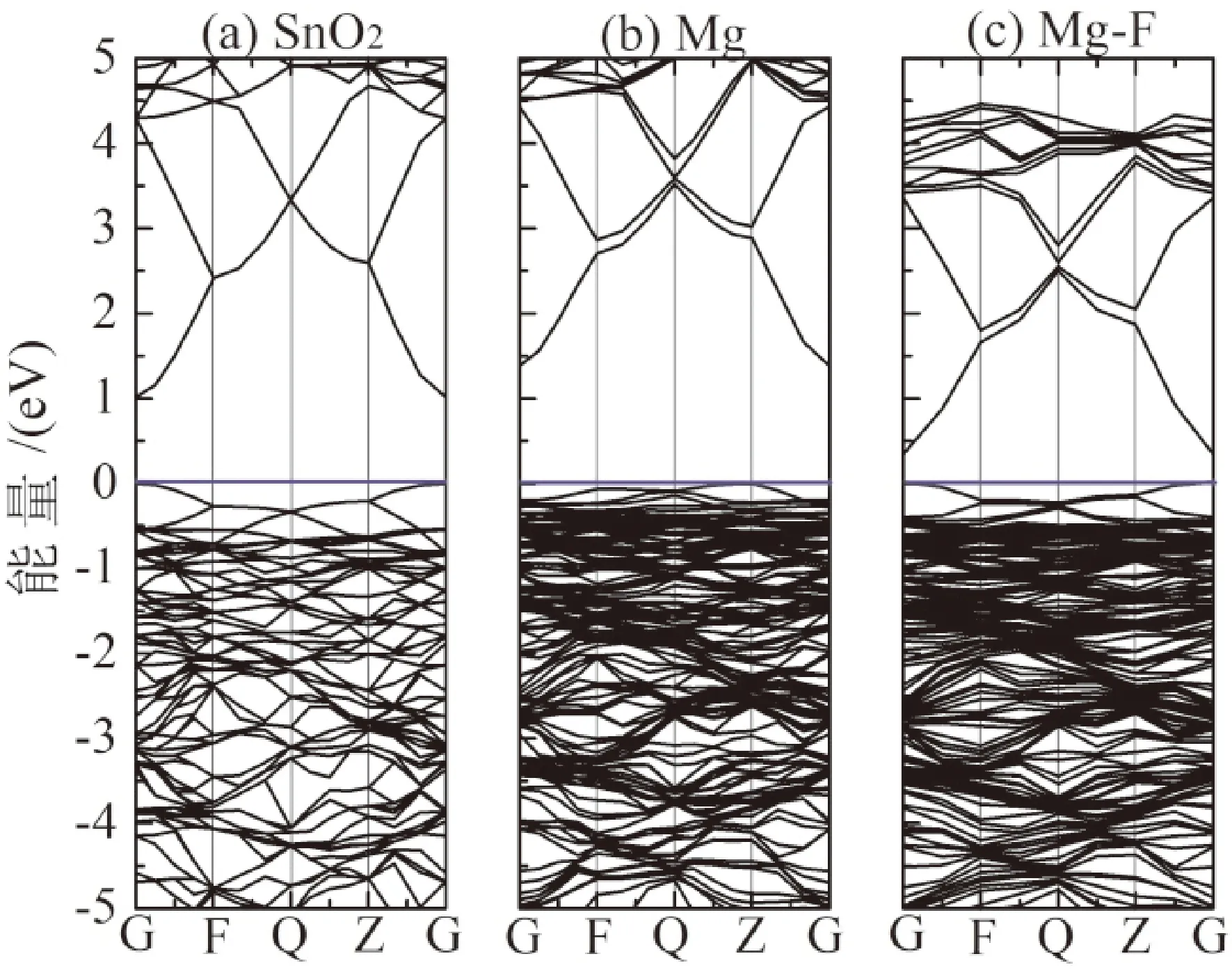
图3 SnO2能带结构图(a)不掺杂;(b)Mg掺杂;(c)Mg-F共掺杂Fig. 3 The band structure of SnO2 (a) un-doped, (b) Mg-doped, (c) Mg-F co-doped
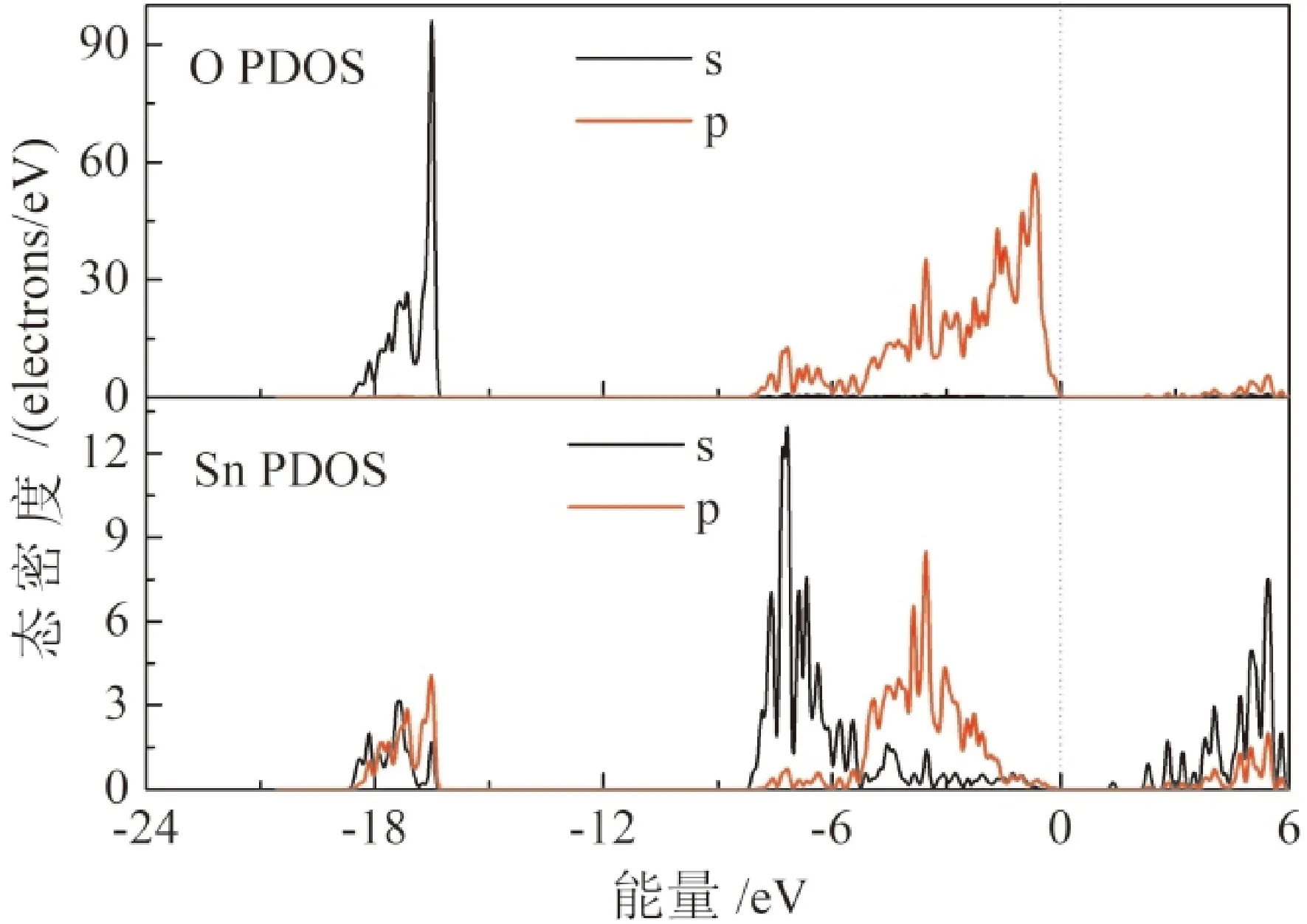
图4 未掺杂时SnO2分波态密度曲线Fig. 4 Partial DOS for undoped SnO2
由图4未掺杂时O、Sn分波态密度图可知,价带分为两大部分,第一部分位于-8.2 eV与费米能级之间,由O 2p和Sn 5s、5p态杂化构成,其中价带顶(VBM)主要由O 2p态杂化构成,2p轨道的局域性会导致空穴载流子局域性强,影响载流子迁移率. 在-18.5 eV— -16.5 eV附近出现的峰值主要由O 2s和Sn 5s、5p态杂化构成. SnO2的导带底(CBM)主要由Sn 5s和5p态杂化构成,SnO2容易形成n型掺杂,且具有较高的电子迁移率.
Mg-F共掺杂时,与未掺杂SnO2相比O分波态密度峰值强度减弱明显,如图5(d)所示. 受Mg 3s电子的影响,Mg分波态密度在-8 eV— 0 eV附近出现峰值,如图5(c)所. Mg 3s轨道与O 2p轨道在价带杂化,在一定程度上降低了O 2p态的局域性,有利于SnO2的p型掺杂. 由图,如图5(b)可知,Mg-F共掺杂虽然也引起Sn分波态密度峰值强度的减弱,在价带顶附近比较Sn 5p、Mg 3s与O 2p态密度图可以看出Sn-O之间的键仍然比Mg-O之间的杂化强. 从图5(a)发现,在价带顶附近F 2p的态密度峰值强度较大,Mg-F之间的键比Mg-O之间的杂化强,那么说明引入施主掺杂元素F有利于Mg受主掺杂的稳定性.
3.3 电荷布居分布
为了分析Mg-F共掺杂SnO2引起的电荷变化,本文计算了Mg-F掺杂SnO2的Mulliken布居分布数据. 表2统计了SnO2未掺杂、Mg单受主掺杂及Mg-F共掺杂时体系的电子重叠布居以及对应键长. 相较于未掺杂体系,Mg-O键之间的键长变化不大,Mg2+与Sn4+半径相近,有利于Mg替换Sn形成受主掺杂. 由表2可知,Sn-O键的离子性较强,Mg单受主掺杂时Mg-O之间电子重叠布居大于Sn-O之间,其值越大说明与Mg-O键的共价性增大. 随着引入F形成施主受主共掺杂体系后,Mg-O1之间电子重叠布居值继续增大,原子之间的重叠增加,共有化增强,可使导电性能进一步提升.
4 结 论
本文采用基于密度泛函理论的第一性原理平面波超软赝势法,计算了Mg-F受主-施主共掺杂SnO2的结构稳定性、能带结构、态密度及布居分布. Mg-F掺杂能形成稳定的掺杂体系,在价带顶附近F 2p的态密度峰值强度较大,Mg-F原子之间杂化强,引入施主掺杂元素F有利于Mg受主掺杂的稳定性,同时Mg-O之间电子重叠布居值增大,原子之间的重叠增加,共有化增强,可使导电性能进一步提升.
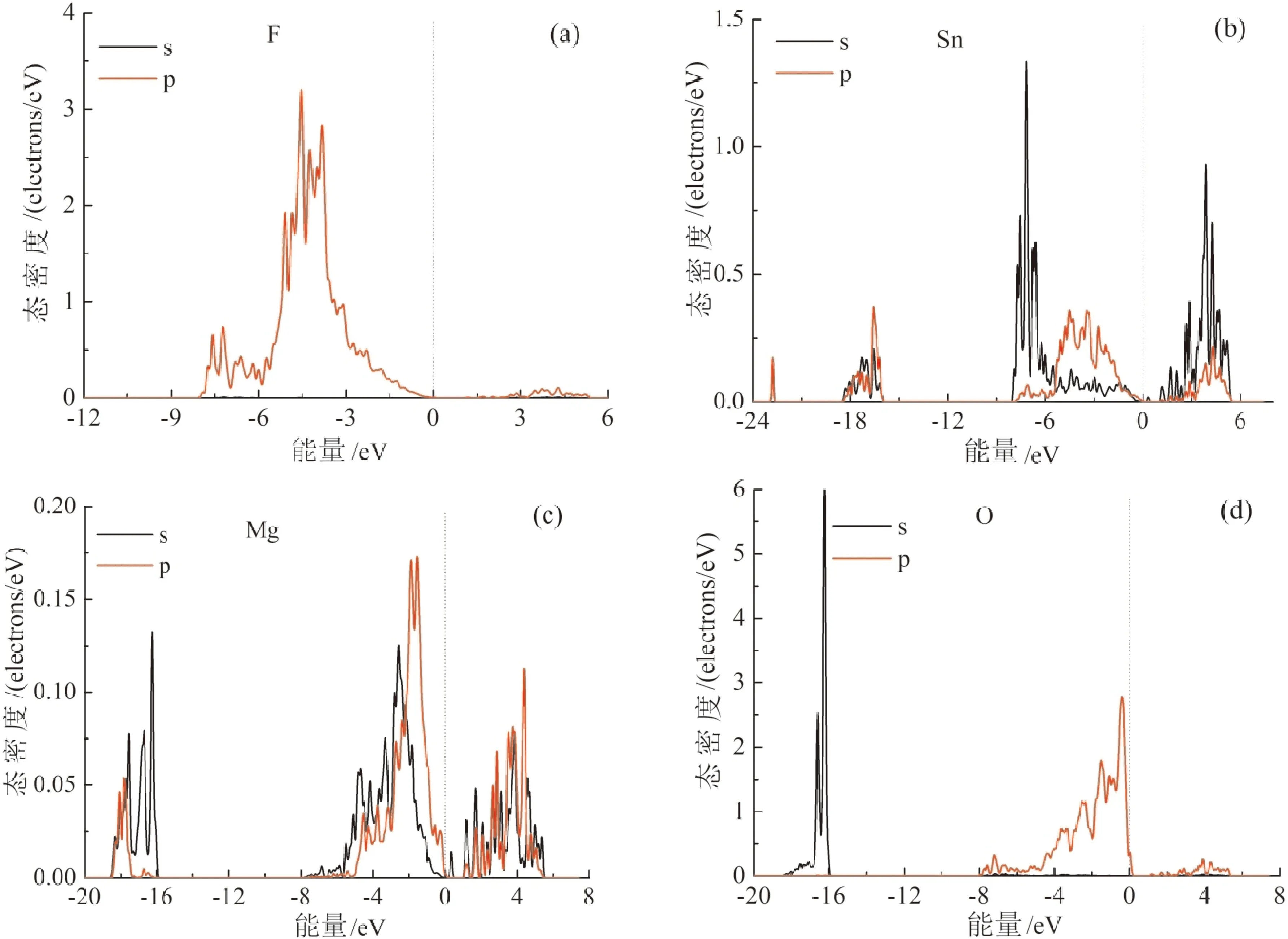
图5 Mg-F共掺杂时分波态密度曲线(a)F;(b)Sn;(c)Mg;(d)OFig. 5 Partial DOS for Mg-F co-doped SnO2 (a) F, (b) Sn, (c) Mg, (d) O
表2 相关原子之间的电子重叠布居和键长
Table 2 Electronic overlapping populations and bond lengths between related atoms
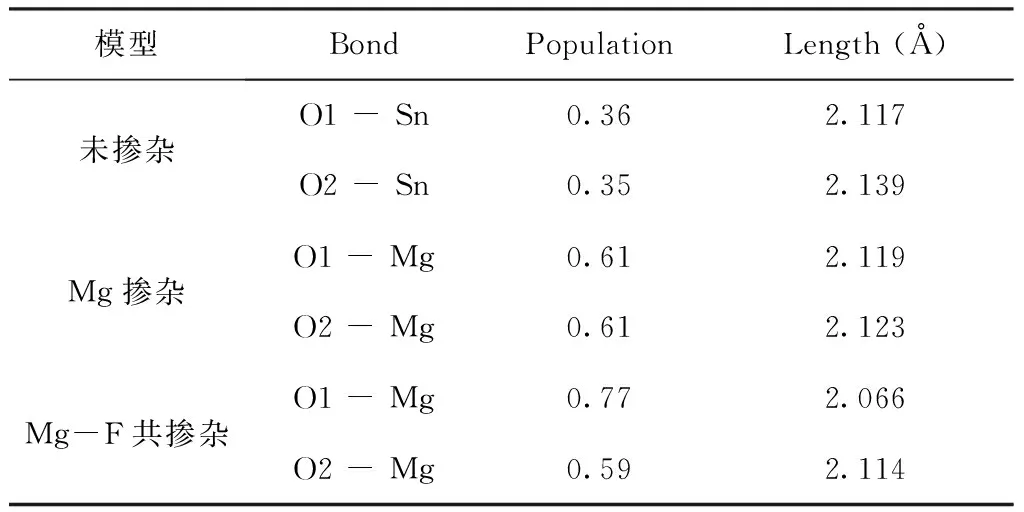
模型BondPopulationLength (Å)未掺杂O1 - Sn0.362.117O2 - Sn0.352.139Mg掺杂O1 - Mg0.612.119O2 - Mg0.612.123Mg-F共掺杂O1 - Mg0.772.066O2 - Mg0.592.114
猜你喜欢
当代作家(2021年11期)2021-12-17
艺术品鉴(2020年6期)2020-12-06
科学(2020年4期)2020-11-26
原子与分子物理学报(2020年3期)2020-05-15
科学(2020年4期)2020-01-11
数学物理学报(2019年5期)2019-11-29
西安工业大学学报(2019年2期)2019-04-02
吉首大学学报(自然科学版)(2018年3期)2018-07-03
领导文萃(2017年6期)2017-03-24
中学生数理化·高一版(2016年7期)2016-12-07
