
运动氧化应激与PI3K-Akt通路研究进展
2019-04-22 06:39吴丽君阮英朝
体育研究与教育 2019年2期
吴丽君,赵 静,孙 卓,阮英朝
机体在正常新陈代谢过程中不断产生自由基,其中约95%为ROS,包括超氧阴离子(.O2)、过氧化氢(H2O2)和羟自由基(.OH)等。它们在细胞内通常可被抗氧化防御系统清除[1]。但是,ROS超量产生、抗氧化体系过负荷将导致机体发生病理生理损伤,被称为氧化应激[2]。有研究表明PI3K-Akt信号通路可抑制氧化应激,但这一研究大多集中在癌症、心血管疾病及糖尿病等慢性疾病类医学领域[3,4],而Akt通路在运动诱导的氧化应激中的作用方式尚不清楚。笔者通过综述运动、氧化应激及Akt相关通路三者间的关系,旨在为更好地认识Akt通路、抑制运动氧化应激及缓解运动疲劳、治疗某些慢性疾病提供参考。
1 PI3K-Akt通路与细胞能量代谢
PI3K通路可以调节细胞增殖、分化、凋亡和应激[5,6]。一些信号蛋白如蛋白激酶B(PKB/Akt)、蛋白激酶C(PKC)和核因子体系(NF-kB)能够识别PI3K并靶向调节下游酶的活性[7]。Akt是PI3K主要的下游靶点,通过跨膜转运及磷酸化后被激活,进而直接或间接调节细胞活动。具体表现为:PI3K通过受体酪氨酸激酶(PTK)或者G蛋白偶联受体(GPCR)启动,之后PI3K主要磷酸化PI4,5P2或PI3,4P2产生PIP3。PIP3分别通过其下游的丙酮酸脱氢酶激酶(PDK1)及mTORC2磷酸化Akt上的Thr308及ser473发挥作用。Thr308及Ser473通常被认为是AKT激活的必要条件[6]。Akt在哺乳动物细胞中存在三种同型AKT1(PKBα)、AKT2(PKBβ)、AKT3(PKBγ),并在调节细胞代谢的过程中发挥作用[8]。
运动过程中能量代谢加剧,PI3K - Akt通路可调节糖代谢及脂肪代谢。胰岛素可通过刺激PI3K-Akt信号通路激活4型葡萄糖转运蛋白(GLUT4)将葡萄糖转运入细胞,从而调节机体糖代谢[9]。Akt通路通过下调过氧化物酶体增殖物激活受体-α(PPARα)及PPARγ辅助激活因子(PGC-1)基因转录而促进脂肪酸氧化[10]。
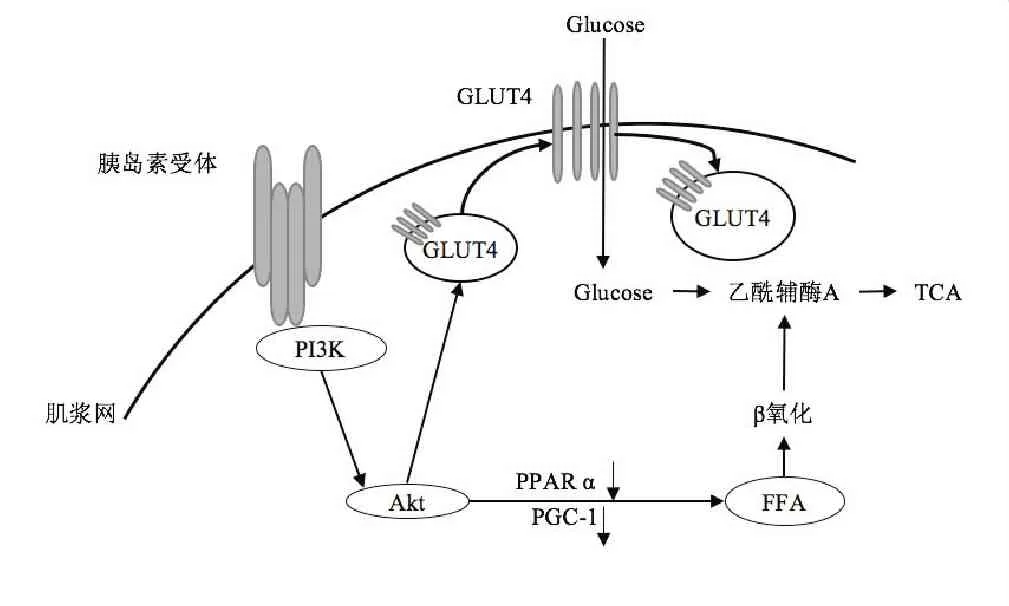
图1PI3K-Akt通路参与调节能量代谢
2 运动诱导氧化应激
细胞氧化还原状态不平衡引起氧化应激,主要原因是ROS超量产生或抗氧化剂活性降低[11~14]。线粒体是有氧呼吸产生ROS的主要部位。线粒体氧化磷酸化过程中电子传递链的电子泄露产生自由基[15],并且几乎所有氧化酶,包括质膜结合的NADPH氧化酶(NOX)、细胞质黄嘌呤氧化酶(XOD)及微粒体细胞色素P450(CYP)等都直接或间接产生ROS[16]。运动过程中骨骼肌细胞能量代谢速率加快,线粒体耗氧量增加导致骨骼肌细胞产生超量ROS[17]。因此运动被认为是诱导机体氧化应激的原因之一。
2.1 不同运动方式对机体产生ROS的影响
运动与氧化应激的关系十分复杂,机体ROS的积累程度取决于运动方式、运动强度及持续时间。有研究表明:中等强度运动产生的少量ROS在细胞内可作为第二信使介导生长因子信号传递[18],而大强度运动导致机体自由基体系紊乱,可引起骨骼肌纤维正常生理环境改变、血管内皮功能障碍[19,20]。还有研究表明剧烈运动干扰氧化还原平衡,导致瞬时性氧化应激。ROS介导的氧化应激在剧烈运动引发的损伤中起关键作用,并且伴随氧化应激的加剧,几种关键的细胞组分,如DNA、蛋白质和膜脂质都受到损伤[21,22]。
2.2 运动氧化应激引起肌肉疲劳及炎症反应
动物实验表明ROS是引起肌肉疲劳的原因之一,在运动至疲劳过程中氧化应激标志物增加[23]。疲劳骨骼肌中各蛋白质和脂质甚至DNA氧化修饰的痕迹都十分明显。氧化应激不仅仅局限于肌肉,在血清及非肌肉组织例如肝脏、肺和中枢神经系统中都存在[24],并且,氧化应激的程度和分布随着运动强度和持续时间的增加而增加,同时过量运动会引起由氧化应激引起的炎症反应,导致受损肌纤维恢复延迟[25]。
抗氧化剂有助于肌肉疲劳恢复。黄芪和丹参提取物改善了小鼠氧化应激指标,增加了小鼠运动耐力[26];ROS清除剂N-乙酰半胱氨酸可延缓兔膈肌疲劳[27];培养基中加入SOD或过氧化氢酶(CAT)可部分抑制氧化应激导致的疲劳[24],抗氧化剂清除ROS还可抑制次最大收缩期间的肌肉疲劳[28,29]。综上所述,运动与氧化应激及疲劳恢复密切相关,抗氧化剂可通过清除超氧化物缓解运动疲劳。
3 运动与Akt磷酸化
3.1 不同运动类型对骨骼肌细胞Akt通路的影响
骨骼肌细胞中,细胞信号转导通路受运动时间、运动方式的影响。在体外实验中,大鼠趾长伸肌(快肌)被动牵拉10分钟后Akt活性显著增加(2倍)。然而,拉伸对大鼠比目鱼肌(慢肌)中的Akt活性没有影响,但应激激活的蛋白激酶p38磷酸化显著。拉伸没有引起糖原合成激酶3(GSK3)靶向位点上的糖原合成酶去磷酸化[30],表明拉伸运动可增加快肌纤维中的Akt磷酸化,但不会引起慢肌中Akt的磷酸化。骨骼肌中Akt对不同类型肌肉的活动产生不同响应。
健康成年男性在空腹状态下进行急性大强度运动后即刻,Akt上的Thr308与Ser473以及Thr37 / 46磷酸化水平降低,表明空腹状态下的高强度运动会抑制Akt 途径,但其具体机制尚不明确[31]。间歇性大强度运动可抑制Akt及其下游效应因子:哺乳动物雷帕霉素靶蛋白(mTOR)和核糖体蛋白S6激酶(p70 S6K)以及Ras-细胞外信号调节激酶(Erk1/2)的活性[32]。在跑步机上进行持续13周,每周5次的有氧运动增加了高血压大鼠Akt磷酸化、减少了GSK3β磷酸化,且有氧运动显著减弱了正常血压组中泛素化蛋白4-羟基壬烯(4-HNE)和脂质过氧化氢的水平[33],表明有氧运动可降低机体ROS水平。Camera研究表明:游泳运动员进行抗阻力运动可激活AMPK通路,举重运动员进行耐力运动可激活Akt通路,而游泳运动员进行耐力运动并不会对Akt通路产生显著影响,可能是由于长期进行某项运动提高了机体对此项运动的适应能力[34]。可见耐力运动和有氧运动刺激Akt磷酸化,长期运动可提高机体对运动的适应能力,运动强度过大反而抑制Akt活性。运动对Akt及其下游效应因子活性的影响虽然已有研究,但不同运动方式对Akt通路的作用机理尚不明确。运动对Akt通路相关蛋白的影响有待深入研究。
大强度运动时机体ROS升高[19~22],ROS含量升高激活Akt抵抗氧化应激[35],与大强度运动抑制Akt活性存在矛盾之处[31,32],有待进一步研究。
3.2 运动刺激Akt对疾病的影响
胰岛素可促进氨基酸进入细胞增加蛋白质合成。有氧运动可通过改善老年人Akt-mTOR信号转导功能来促进肌肉蛋白合成对胰岛素的响应[36]。衰老Wistar大鼠连续五周进行短时间(4—6分钟/天)、低强度跑台运动,可增加其比目鱼肌和心肌氧耗量,并可逆转在水迷宫评估中与年龄相关的长期空间学习和记忆障碍。这种老龄大鼠认知功能的增强伴随着Akt与cAMP反应元件的结合(CREB),最终显著增加了老龄大鼠海马神经营养因子(BDNF)mRNA表达及其蛋白水平的变化[37]。这一结果表明长期进行低强度锻炼可以通过Akt等信号通路改善老龄大鼠的认知和突触可塑性。另外ST大鼠每天游泳1小时可通过激活缺氧诱导因子(HIF-1α)、血管内皮生长因子、基质金属蛋白酶-2途径诱导Akt活化来改善老龄大鼠缺氧反应中新血管的形成[38]。可见,有氧运动可通过激活Akt改善机体骨骼肌、心血管及神经系统功能。
4 氧化应激与Akt信号转导通路
ROS少量升高时可作为信号激活细胞内下游Akt、丝裂原活化蛋白激酶(MAPK)、AMP依赖的蛋白激酶AMPK、蛋白激酶C(PKC)等蛋白激酶,以增加氧化应激的缓冲能力[39]。当机体ROS大量升高时,机体氧化还原平衡被打破,激活细胞Akt通路抵抗氧化应激损伤[35]。
PI3K-Akt通路在氧化应激时可调节细胞凋亡[4]。有研究表明激活PI3K-Akt通路可以保护神经细胞免受氧化应激的损伤[40]。抑制Akt活性使细胞对ROS更加敏感,更易受到氧化损伤[41]。PI3K-Akt信号传导在包括血红素加氧酶1(HO-1)、谷氨酸半胱氨酸连接酶(GCL)在内的抗氧化酶表达中发挥重要作用[42,43]。因此Akt相关信号转导通路是机体抵抗氧化应激的重要通路之一。
4.1 抗氧化药物对Akt通路的影响
许多抗氧化药物在细胞内通过Akt通路发挥作用。Apicella等人研究表明补充甜菜碱可增强急性运动后人体Akt磷酸化导致蛋白质合成增加[44]。白藜芦醇可通过抑制氧化应激并激活Akt通路产生更有效的信号转导改善人体胰岛素敏感性[45]。新型合成硫化氢(H2S)、释放型丹参素(SDSS)通过抑制氧化应激,激活PI3K-Akt通路保护MC3T3-E1成骨细胞避免H2O2诱导的细胞凋亡[46]。高密度脂蛋白也可通过激活PI3K - Akt途径保护间充质干细胞避免氧化应激诱导的细胞损伤[47]。
人体和动物试验均证实抗氧化药物可作用于Akt通路抵抗氧化应激从而改善机体机能,为部分疾病的治疗指明了方向。
4.2 PI3K-Akt通路通过作用于Nrf2抵抗氧化应激
研究表明:PI3K-Akt信号通路作用于核因子红细胞相关因子2(Nrf2)能够使细胞免受氧化应激和炎症反应的损害[3,48,49]。Nrf2是碱性区域亮氨酸拉链(bZip)转录因子的cap′n′collar(CNC)亚族的成员,属于抗氧化调节剂。Nrf2与抗氧化应答元件(ARE)结合启动下游包括过氧化氢酶 ( CAT )、超氧化物歧化酶(SOD)、NADPH醌氧化还原酶1(NADPH NQO1)、谷胱甘肽S-转移酶(GST)和血红素加氧酶1(HO-1)等一系列抗氧化酶和II解毒酶基因的转录[50]促进抗氧化酶的表达、清除过剩自由基,并将体内外源和内源性有毒物质转化为无害的代谢产物排出体外[51]。
Nrf2在细胞质中与凯洛奇样ECH相关蛋白1(Keap1)结合通过范素化作用保持在低水平状态[52]。氧化应激发生时,ROS首先激活PI3K-Akt,随后Akt抑制糖原合成酶激酶3β(GSK3β)。GSK3β是PI3K-AKT信号传导途径的主要下游靶点。许多研究提出Akt磷酸化介导GSK3β-Nrf2通路调节细胞中的抗凋亡和抗氧化功能[35,53,54]。Akt激活后抑制GSK3β的活性,导致p21cip激活,同时Keap1通过识别细胞的氧化还原状态并修饰其半胱氨酸残基与Nrf2分离,p21cip代替 Keap1与Nrf2结合[41]。随后,Nrf2移动到核内并通过与抗氧化应答元件(ARE)结合反式激活HO-1、还原型谷胱甘肽(GSH)等靶基因以抑制氧化应激[55]。HO-1是Nrf2靶基因的代表之一。其特点是能够逆转氧化应激和损伤[56]。GSH是体内的主要抗氧化剂之一,在氧化应激状态下,GSH被氧化为氧化型谷胱甘肽(GSSG)进而消除ROS。
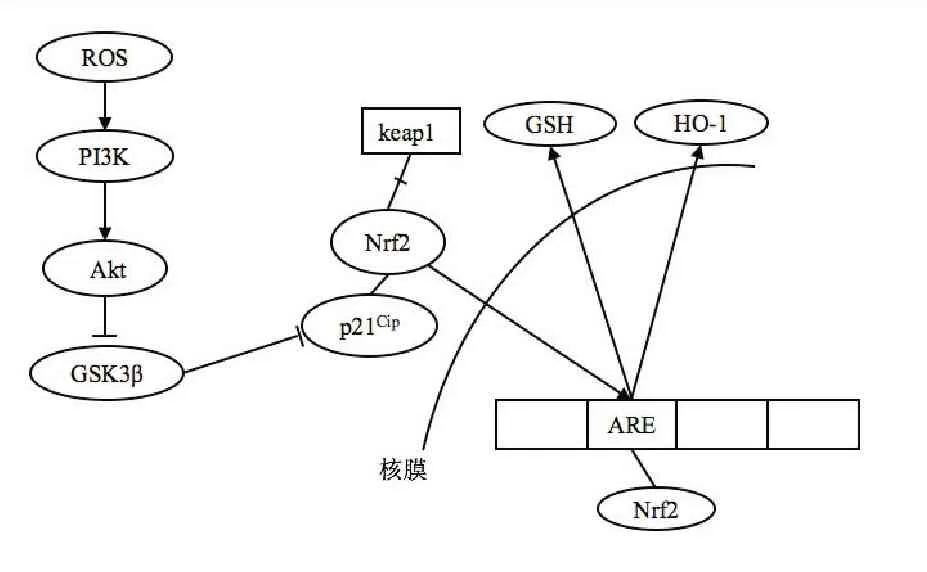
图2Akt-Nrf2抗氧化通路
熊去氧胆酸(UDCA)通过激活PI3K-Akt-Nrf2途径促进合成GSH,从而降低肝炎细胞氧化应激水平[57];紫杉醇通过激活PI3K-Akt上调Nrf2调节II解毒酶,从而保护ARPE-19细胞免受氧化应激损伤[43]。磷酸肌酸(PCr)在体内外的神经保护作用依赖于通过Akt介导的Nrf2-HO-1途径使线粒体功能正常化并且降低细胞氧化应激水平[58]。
综上所述,PI3K-Akt-Nrf2通过上调机体抗氧化酶抵抗氧化应激,但是PI3K-Akt通路只是机体诸多抗氧化通路的一种,Nrf2也受AMPK通路调节[59]。PI3K-Akt-Nrf2在机体抗氧化应激过程中的重要性有待进一步研究。
6 小结
大负荷运动导致机体超量产生ROS ,ROS是引起肌肉疲劳的原因之一。PI3K-Akt通路参与调节细胞能量代谢,也参与细胞抗氧化应激。ROS可刺激Akt磷酸化,从而激活其下游Nrf2参与机体抗氧化应激反应。由于细胞中其他信号转导通路如AMPK等也通过刺激Nrf2抵抗氧化应激,故Akt通路在抗氧化应激的过程中是否起主要作用尚不明确。大强度运动抑制骨骼肌细胞中Akt磷酸化,但是其作用机制尚不明确。笔者总结氧化应激、运动及Akt之间的关系,可为深入研究运动氧化应激提供全新的视角,为氧化应激相关疾病的预防、运动疲劳的恢复等研究提供参考。
猜你喜欢
基层中医药(2022年4期)2022-07-22
体育科技文献通报(2022年3期)2022-05-23
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
医学综述(2021年16期)2021-12-01
科学(2020年2期)2020-08-24
三农资讯半月报(2020年3期)2020-03-09
山西农业大学学报(自然科学版)(2020年1期)2020-03-04
运动(2018年14期)2018-07-16
分析化学(2017年12期)2017-12-25
