
脊髓损伤机制研究的生物信息学分析
2019-04-17 08:45任恩惠张广智贺学岗高一诚杨风光马占军解琪琪康学文
生命科学研究 2019年6期
任恩惠,张广智,贺学岗,高一诚,杨风光,杨 亮,马占军,解琪琪,汪 静,康学文*
(1.兰州大学第二医院骨科,中国甘肃兰州730000;2.兰州大学第二临床医学院,中国甘肃兰州730000)
脊髓损伤(spinal cord injury,SCI)常继发于交通事故、坠落伤、运动损伤等原因导致的椎体骨折[1~2],并且通常会造成瘫痪等严重的后果。脊髓损伤后损伤部位难以修复,患者生活质量低,是目前临床上难以解决的医学难题。脊髓损伤可分为原发性脊髓损伤和继发性脊髓损伤。原发性脊髓损伤常造成神经不可逆损伤,并且由于损伤部位往往伴随脊髓水肿、脊髓组织缺血、过氧化自由基生成增多以及炎症反应等症状,因此可引起继发性脊髓损伤[3~6]。由此可知,对于脊髓损伤发展过程中分子机制的探究,可以预测继发性脊髓损伤潜在的治疗靶点。然而,目前关于脊髓损伤发展过程中分子机制的研究仍然不清楚。随着生物医学的发展,研究者可通过高通量基因芯片技术探究疾病发生发展过程中分子的变化,这为脊髓损伤发展机制的研究提供了新的方法。
本文通过生物信息学分析,对脊髓损伤中具有表达差异的基因进行功能富集,分析得到了参与脊髓损伤发展过程的重要分子和通路,为脊髓损伤发展机制的研究提供了新的思路,同时也为脊髓损伤后修复的研究提供了新的潜在靶点。
1 材料和方法
1.1 芯片数据信息
本文分析的基因芯片数据均来自于GEO数据库,分析的数据集为GSE42828、GSE47681和GSE-5296,测序平台为GPL1261[Mouse430_2]Affymetrix Mouse Genome 430 2.0 Array。样本来源于Mus musculus。提取3个数据集中样本的基因芯片数据,并将所有的样本分为脊髓损伤组(SCI组)和正常组(normal组)。样本具体分布情况如表1所示。
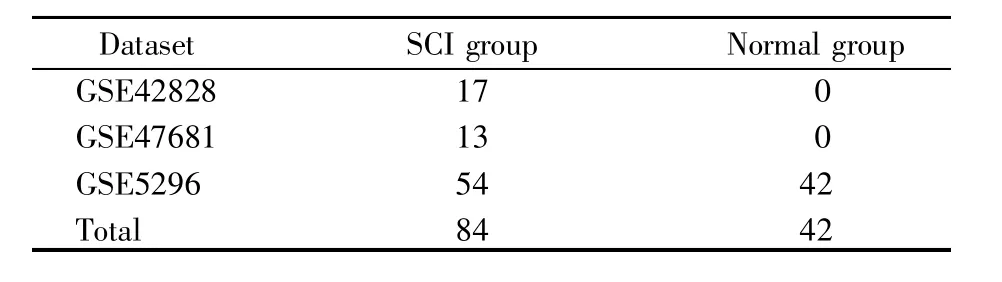
表1 数据集中的样本分布Table 1 Sample distribution in the dataset
1.2 基因芯片数据的标准化以及差异基因的筛选
合并下载的基因芯片数据,并通过R语言中的sva包处理来自3个不同数据集样本之间的批次效应。对合并后的数据进行标准化,并通过主成分分析(principal component analysis,PCA)监测标准化后样本的分布情况。用R语言中的limma包处理标准化后的基因芯片数据,得到差异基因[7],差异基因筛选条件为:|log2(fold change)|>0.9,P-value<0.05。
1.3 功能富集分析
DAVID数据库可从生物过程(biological process,BP)、细胞组分(cellular component,CC)和分子功能(molecular function,MF)3个方面注释差异基因的功能,是目前进行差异基因功能注释的权威数据库[8~9]。我们利用DAVID数据库对分析得到的差异基因进行GO(gene ontology)分析。同时,利用KEGG(Kyoto Encyclopedia of Genes and Genomes)数据库[10](https://www.kegg.jp/)对差异基因进行通路富集分析,从而得到参与脊髓损伤发展过程的重要通路。
1.4 蛋白质相互作用网络构建、hub基因获取及其功能分析
STRING数据库是一个可以构建蛋白质间相互作用网络的数据库[11~12]。我们利用STRING构建蛋白质相互作用(protein-protein interaction,PPI)网络,并通过Cytoscape软件(http://cytoscape.org/download_old_versions.html)进行可视化。随后,利用Cytoscape中的cytoHubba插件经MCC算法将蛋白质调控网络中得分最高的10个基因定义为hub基因,并用箱式图展示了hub基因在SCI组和normal组中的表达情况。GeneCards数据库(https://www.genecards.org/)是一个综合性数据库,通过该数据库能够查询基因的功能。我们通过GeneCards数据库查询得到hub基因的主要功能。
1.5 GSEA分析
GSEA(gene set enrichment analysis)[13]可对表达差异不明显的基因进行功能注释,从而补充差异基因筛选过程中遗漏的关键基因。我们通过R语言clusterProfiler包对基因表达数据进行GSEA分析,进一步完善了生物信息学分析的结果。
2 结果
2.1 数据质控和差异基因的筛选
从GEO数据库下载GSE42828、GSE47681和GSE5296三个数据集中的CEL文件,对不同数据集中基因芯片数据的批次效应进行处理,结果如图1所示,可见批次效应去除成功。进一步对处理后的芯片数据进行标准化处理,结果如图2所示,可见标准化后的数据可用于后续分析。PCA结果表明数据合并之后SCI组和normal组中的样本能够分开(图3)。对标准化后的基因芯片数据进行贝叶斯检验,总共得到104个上调的差异基因。图4以火山图的形式展示了所有基因的分布情况,并标注了hub基因所处的位置。
2.2 GO分析和KEGG分析
用DAVID数据库对得到的104个差异基因进行GO分析,结果如表2所示。差异基因主要参与炎症反应、中性粒细胞趋化性、细胞对白细胞介素-1的反应、星形胶质细胞迁移和血管生成等生物过程。就细胞组分而言,差异基因主要分布在细胞外区域、细胞表面、细胞外外泌体、质膜、细胞内膜结合细胞器和内质网等部位。就分子功能而言,差异基因主要与葡糖醛酸基转移酶活性、趋化因子活性、细胞因子活性和细胞因子受体活性等相关。KEGG分析结果表明,脊髓损伤后TNF信号通路、NF-κB信号通路、趋化因子信号通路和NOD样受体信号通路等相关通路激活(表2),提示以上通路在脊髓损伤发展过程中发挥重要的作用。
图1 批次效应处理图Fig.1 Batch effect processing diagram

图2 原始数据标准化图Fig.2 Raw data normalization map

图3 主成分分析结果Fig.3 Principal component analysis results
2.3 PPI网络和hub基因
将104个差异基因导入STRING数据库,分析得到其所编码蛋白质之间的相互作用关系(图5A)。随后,通过Cytoscape中的cytoHubba插件分析得到TYROBP、ITGB2、PTPRC、FCER1G、C3AR1、C1QB、CYBB、FCGR2B、PLEK 和 MPEG1等 10个 hub基因(图5B)。10个hub基因在SCI组和normal组中的表达如图6所示,由图可知所有hub基因在SCI组的表达明显高于normal组。进一步通过Gene-Cards数据库查询得到每一个hub基因的主要功能,结果提示炎症反应在脊髓损伤发展过程中发挥着重要的作用(表3)。

图4 差异基因表达情况分布红色的点代表符合筛选标准的差异基因的分布,灰色的点代表差异基因以外其他基因的分布。Fig.4 Differential gene expression distributionRed dots represent the distribution of differential genes that meet the screening criteria,and grey dots represent the distribution of genes other than the differential genes.
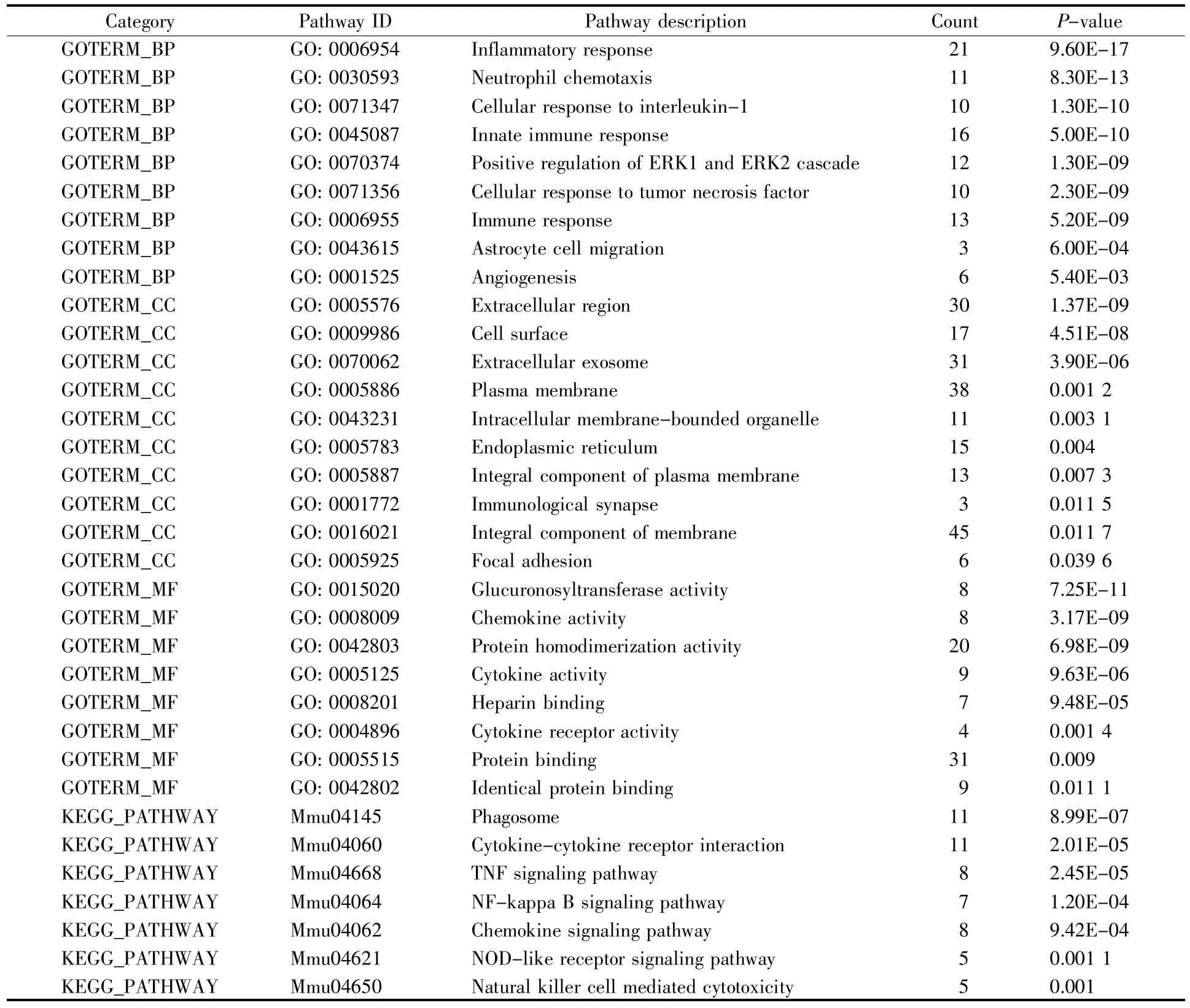
表2 GO和KEGG分析结果Table 2 GO and KEGG analyses
2.4 GSEA分析
通过对基因表达数据进行GSEA分析,得到图7所示的结果。从图中可以看出,CCL2、FOS、CDK1、CCND1、IL1B和BAX等基因在脊髓损伤的发展过程中可能发挥着重要的作用,同时IL-17信号通路和p53信号通路可能参与了脊髓损伤的发展机制。
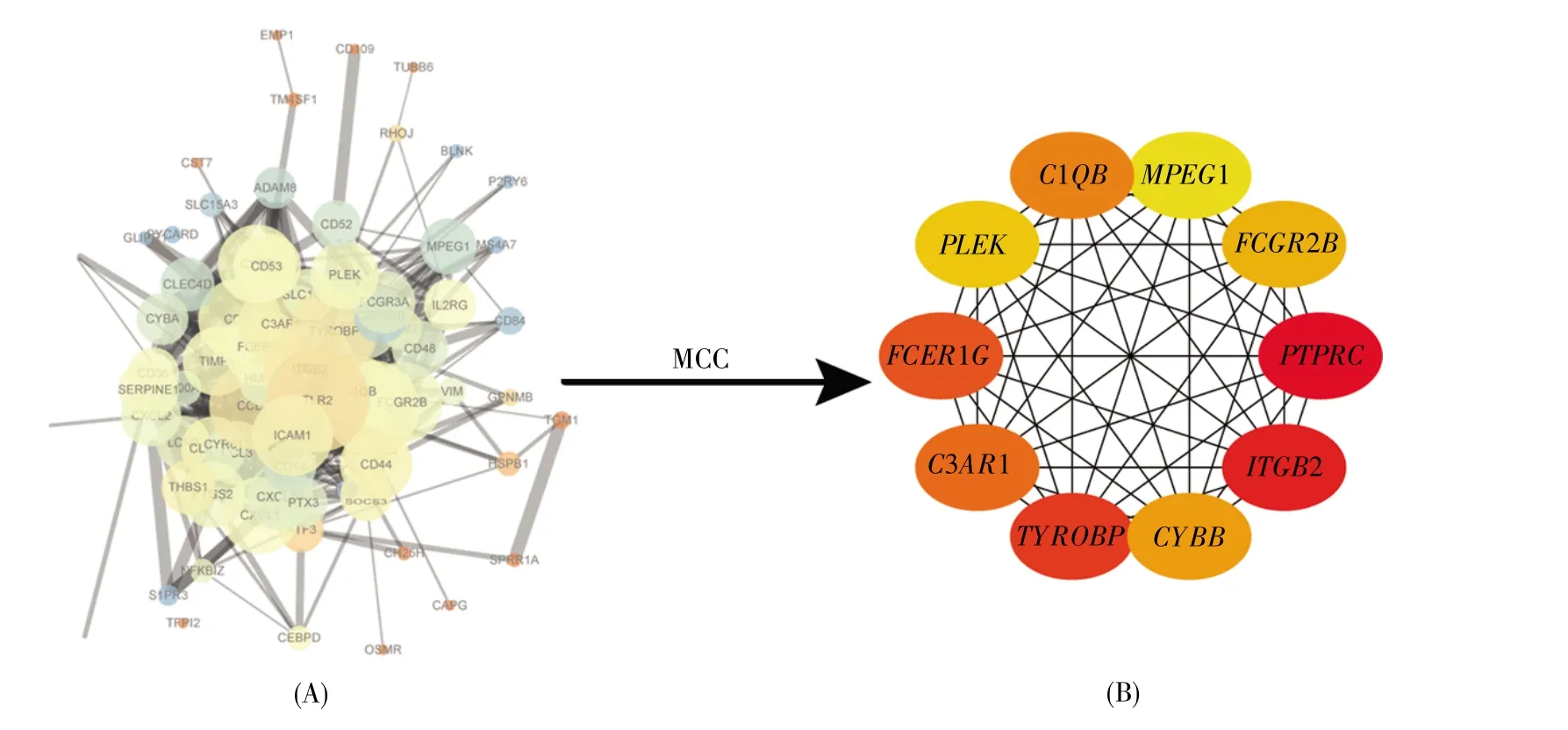
图5 PPI网络和hub基因(A)蛋白质相互作用网络。节点的大小代表度,颜色深浅代表聚类系数,线条的粗细代表综合评分;(B)Hub基因相互作用网络。颜色由红到黄表明基因得分由高到低。Fig.5 PPI network and hub genes(A)Protein interaction network.The size of the circle represents the degree,the depth of the color represents clustering coefficient,and the size of line represents the combined score;(B)Hub gene interaction network.Colors from red to yellow indicate that the gene scores are from high to low.
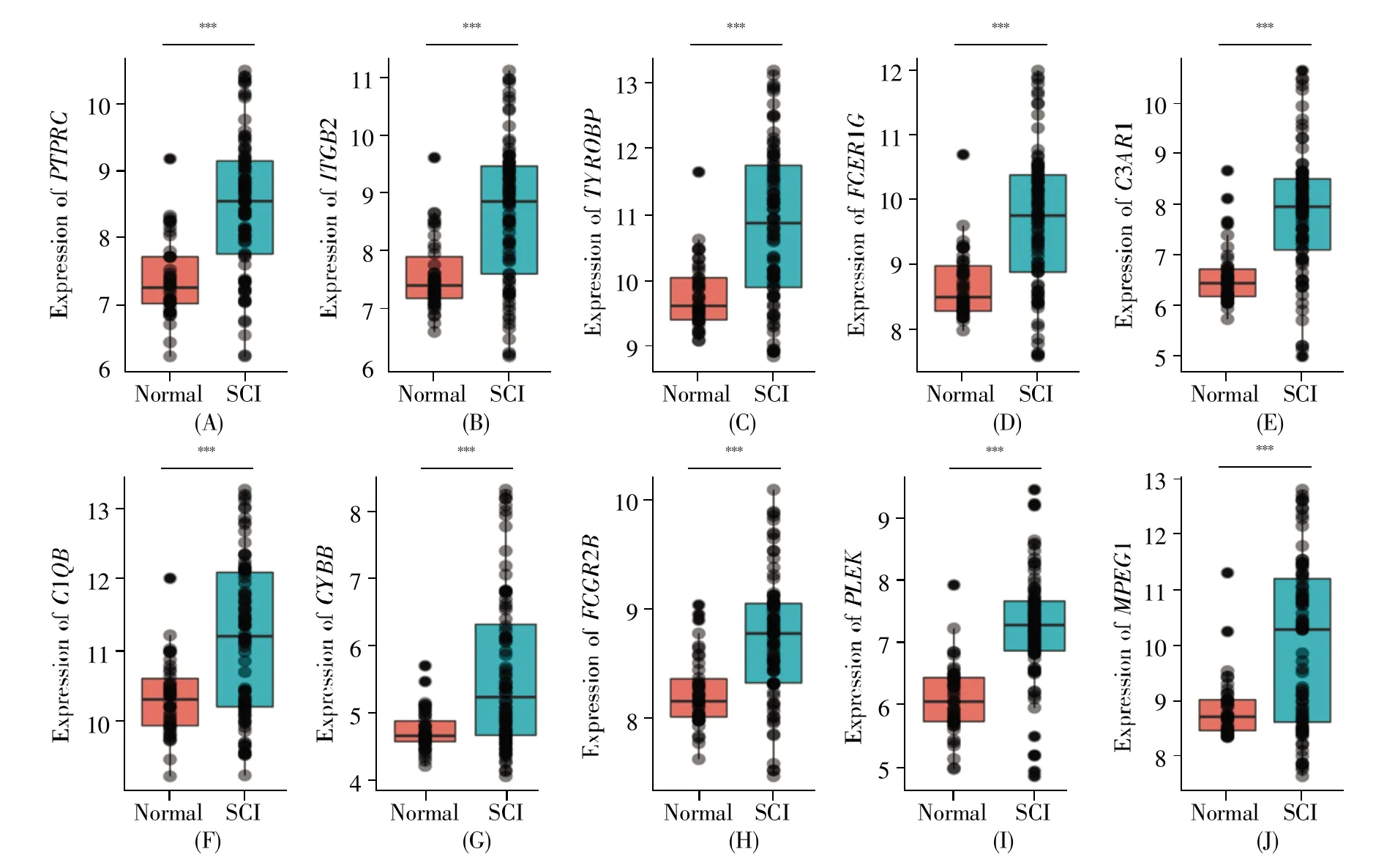
图6 Hub基因表达情况Fig.6 Hub gene expression map
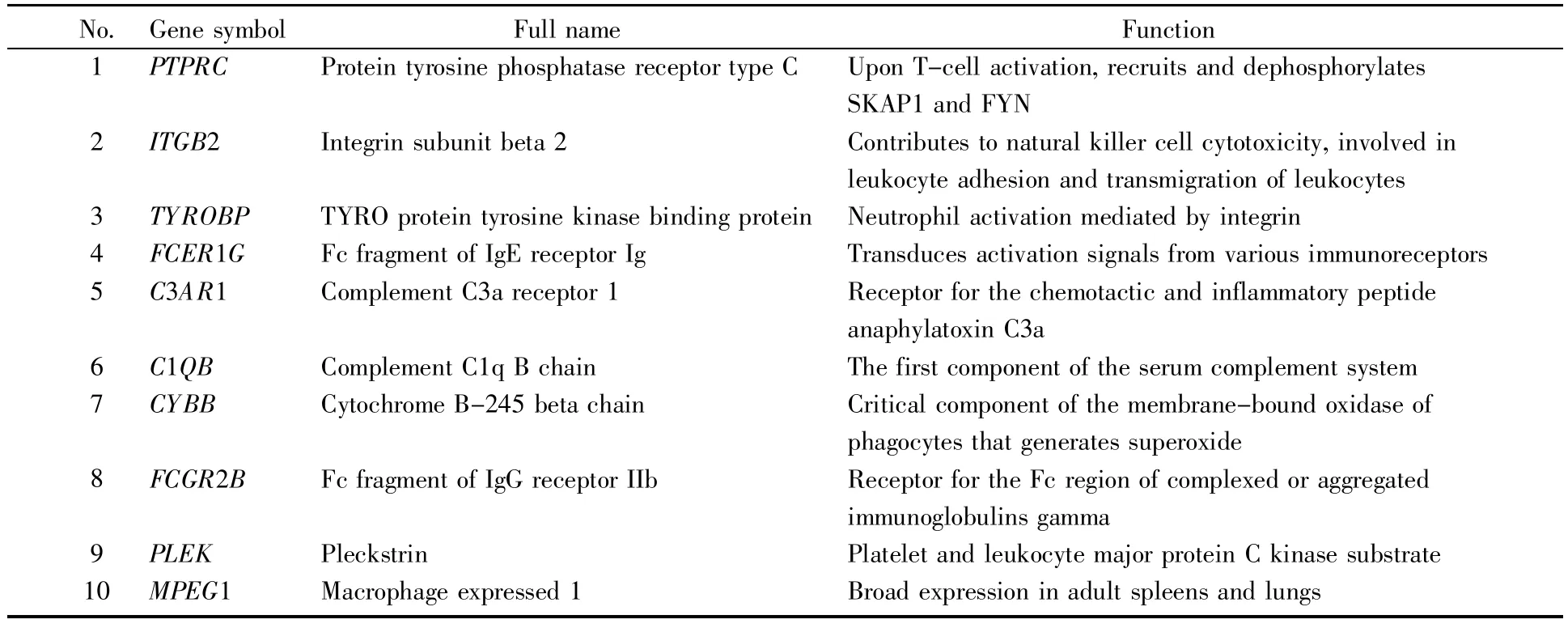
表3 Hub基因的功能Table 3 Functions of the hub genes
4 讨论
急性脊髓损伤常常导致患者损伤节段以下感觉障碍、运动功能障碍、大小便失衡等[14],严重影响患者的生活质量。Liu等[15]总结了脊髓损伤后的病理机制:脊髓损伤后机体产生的炎症反应、活性氧和氧化应激能够引起神经细胞凋亡等继发性损伤,而局部的神经胶质细胞可通过分泌神经营养因子产生代偿性的保护作用。脊髓损伤后,中性粒细胞、巨噬细胞和淋巴细胞等炎性细胞聚集在损伤部位,并通过分泌肿瘤坏死因子-α(TNF-α)、白细胞介素-1β (IL-1β)、激活蛋白-1(AP-1)和核因子κB(NF-κB)等炎性因子介导继发性脊髓损伤[16~20]。氧化应激导致的损伤机制在脊髓损伤发展过程中发挥着重要的作用,在脊髓损伤后的数小时内,过氧化氢(H2O2)、超氧化物和羟基自由基等活性氧在损伤部位聚集,对脊髓中的蛋白质、脂质和DNA造成氧化损伤,从而对脊髓造成二次损伤[21~22]。相关研究表明,脊髓损伤不仅会导致已经损伤的细胞产生凋亡,而且会介导损伤部位正常的神经细胞产生凋亡[23~24]。目前,虽然已有大量的研究对脊髓损伤分子机制进行了探究,但是相关工作仍然处于探索阶段,很难为临床治疗提供指导。因此,针对脊髓损伤后相关分子机制的研究,不仅可加强人们对脊髓损伤发展机制的认识,也可为临床治疗提供新的思路。基于此,本文对不同数据集中的基因芯片数据进行了合并,分析得到了104个高表达的差异基因。
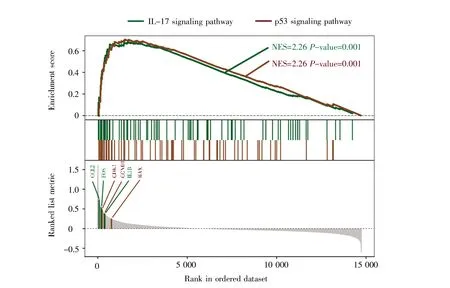
图7 GSEA分析结果Fig.7 GSEA analysis results
功能富集结果表明,104个差异基因主要位于细胞外基质,主要参与了炎症反应、中性粒细胞趋化性、星形胶质细胞迁移和血管生成等过程。我们通过对蛋白质相互作用网络展开分析,得到10个 hub 基因:TYROBP、ITGB2、PTPRC、FCER1G、C3AR1、C1QB、CYBB、FCGR2B、PLEK 和 MPEG1。其中TYROBP、PTPRC和C1QB在脊髓损伤发展机制的研究中有报道[25~28],但是关于这些分子的具体调控机制仍不清楚。C1QB为补体系统中的重要成分,相关研究表明,该分子参与了脊髓损伤后补体系统的激活,从而造成神经元的继发性损伤[29~31]。除此之外,C1QB也参与了小胶质细胞介导继发性损伤的过程[32]。目前,关于ITGB2、FCER1G、C3AR1、CYBB、PLEK和MPEG1等基因在脊髓损伤发展机制中的研究很少有报道,本文的hub基因筛选结果为脊髓损伤发展机制的探究提供了新的思路。
KEGG分析结果显示,TNF信号通路、NF-κB信号通路、趋化因子信号通路和NOD样受体信号通路等在脊髓损伤部位激活。相关研究表明,TNF信号通路不仅参与脊髓损伤后的炎症反应,也与脊髓损伤后神经细胞的凋亡有关,提示TNF信号通路在脊髓损伤发展过程中有着重要的作用,阻断该通路可能有助于继发性脊髓损伤的治疗[33~34],这与我们预测的结果相似。近年来,相关研究报道,NF-κB信号通路、趋化因子通路、NOD样受体信号通路、自然杀伤细胞介导的细胞毒性等信号通路在脊髓损伤发展机制中有着重要的作用[35~37],然而关于其机制的具体情况仍不清楚,很难为临床治疗提供指导,因此对这些通路展开深层次的探究将有助于进一步加深人们对脊髓损伤发展机制的认识。
为了补充差异基因筛选的不足,我们对所有基因的表达数据进行了GSEA分析。通过GSEA分析预测得到 CCL2、FOS、CDK1、CCND1、IL1B 和BAX等关键基因,也预测到IL-17信号通路和p53信号通路。趋化因子CCL2是神经损伤后释放的一种损害性因子,主要参与局部炎症和疼痛机制[38]。在脊髓损伤后,局部神经细胞可通过高表达CCL2,促进损伤部位的炎症反应,从而降低神经重塑的可能性,导致损伤的脊髓难以恢复[38~39]。FOS是一种疼痛刺激脊髓高表达的分子[40~41]。相关研究表明,神经损伤后,表达cFOS的神经元明显减少[41~43],说明该分子可能参与脊髓损伤后的保护机制。CDK1主要参与脊髓损伤过程中的凋亡机制,抑制该分子可明显减少神经细胞的凋亡,从而抑制继发性脊髓损伤的发展,促进脊髓损伤后的修复[44~45]。IL-1参与脊髓损伤后炎症反应的机制早已被证明[46~47]。BAX是凋亡机制的标志分子,大量文献证明凋亡机制在脊髓损伤发展过程中有重要的作用。综上所述,CCL2、FOS、CDK1、IL1B和BAX等基因在脊髓损伤发展机制中有重要的作用,这与我们的GSEA分析结果一致。而CCND1在脊髓损伤中的作用仍未证明,这为脊髓损伤机制的探究提供了新的方向,有助于发现脊髓损伤新的治疗靶点。相关研究表明,IL-17信号通路参与了脊髓损伤后的炎症反应,从而可以促进继发性脊髓损伤,并抑制脊髓损伤后的修复[48~49]。p53信号通路是肿瘤研究中的明星通路,我们通过GSEA分析,证明该通路在脊髓损伤发展过程中可能发挥重要的作用,但是其具体作用机制仍有待进一步的研究。
综上所述,我们通过生物信息学的方法综合分析了不同数据集中的基因芯片数据,成功地预测 了 TYROBP、ITGB2、PTPRC、FCER1G、C3AR1、C1QB、CYBB、PLEK、MPEG1、CCL2、FOS、CDK1、CCND1、IL1B和BAX等有可能在脊髓损伤发展机制中产生重要作用的分子,同时也预测到TNF信号通路、NF-κB信号通路、趋化因子信号通路、NOD样受体信号通路、IL-17信号通路和p53信号通路等可能与脊髓损伤发展机制关系密切,为脊髓损伤机制的研究提供了生物信息学支持。当然,关于这些通路的具体机制,仍然需要大量的生物实验进行验证,这样才能为我们对脊髓损伤发展机制的认识提供坚实有力的证据。
猜你喜欢
军事文摘(2022年8期)2022-11-03
今日农业(2021年4期)2021-06-09
知识文库(2020年5期)2020-05-11
心电与循环(2020年1期)2020-02-27
江苏农业科学(2017年5期)2017-04-15
现代检验医学杂志(2016年4期)2016-11-15
中国运动医学杂志(2016年3期)2016-07-10
湖北农业科学(2014年3期)2014-07-21
中国中医药现代远程教育(2014年21期)2014-03-01
中国中医药现代远程教育(2014年20期)2014-03-01
