
胆汁酸和肠道菌群相互作用对非酒精性脂肪性肝病的影响
2019-04-17 08:45:12杜传德张晓晨李宗杰
生命科学研究 2019年6期
杜传德,张晓晨,李宗杰
(1.临沂市河东区疾病预防控制中心,中国山东临沂276034;2.临沂市河东区人民医院,中国山东临沂276034;3.中国农业科学院上海兽医研究所,中国上海200241)
胆汁酸(bile acids,BAs)是由肝细胞以胆固醇为原料在胆固醇7α羟化酶(cholesterol 7α-hydroxylase,CYP7A1)的催化下合成的一类小分子物质,不仅参与脂类物质的消化、吸收,而且还可作为重要的信号调节因子影响能量代谢、炎症反应以及肝脏疾病的发生和发展。最新研究发现,胆汁酸与肠道菌群的相互作用和对话交流与非酒精性脂肪性肝病(non-alcoholic fatty liver disease,NAFLD)有密切关系[1]。人体肠道中数量庞大的肠道菌群能产生维生素、短链脂肪酸(short-chain fatty acids,SCFAs)、脂多糖(lipopolysaccharide,LPS)和内生性乙醇(endogenous ethanol)等代谢产物,这些细菌产生的营养物质和有毒物质被肠道吸收后经肠门静脉供给肝脏,在肝脏内被代谢后会影响脂代谢相关基因的转录活性,并可能诱导免疫细胞产生多种细胞因子,从而引起炎症反应[2]。因此,肠道菌群及其代谢产物通过肠-肝轴(gut-liver axis)影响人体的免疫稳态、炎症感染和患病风险[3]。
在肝细胞内合成的初级胆汁酸包括胆酸(cholic acid,CA)、鹅脱氧胆酸(chenodeoxycholic acid,CDCA)、牛磺胆酸(taurocholic acid,TCA)、牛磺脱氧胆酸(taurochenodeoxycholic acid,TCDCA)、甘氨胆酸(glycocholic acid,GCA)和甘氨脱氧胆酸(glycochenodeoxycholic acid,GCDCA)。进食后胆囊收缩将初级胆汁酸排入肠道,进入肠道的胆汁酸可以乳化脂肪并促进脂溶性物质的消化与吸收。绝大多数初级胆汁酸在回肠末端以结合胆汁酸形式被主动重吸收,其余少量胆汁酸经肠道菌群产生的胆酸盐水解酶(bile salt hydrolase,BSH)的去结合作用形成游离胆汁酸,随后发生脱羟基作用形成次级胆汁酸,如脱氧胆酸(deoxycholic acid,DCA)、石胆酸(lithocholic acid,LCA)和熊去氧胆酸(ursodeoxycholic acid,UDCA)等。大多数次级胆汁酸可以在回肠内重吸收并经门静脉系统回到肝脏[4~5]。与正常人相比,非酒精性脂肪性肝病(NAFLD)患者的胆汁酸池大小以及胆汁酸成分均发生了显著改变,而且血清中的成纤维细胞生长因子-19(fibroblast growth factor-19,FGF-19)水平更低,表明胆汁酸代谢异常可影响非酒精性脂肪性肝病(NAFLD)的发生和发展[6]。
1 胆汁酸代谢与肠道菌群的对话交流机制
1.1 肠道菌群对胆汁酸代谢的影响
肝细胞中的胆汁酸合成是由细胞色素P450介导的胆固醇氧化反应,主要通过经典途径和替代途径生成胆酸(CA)和鹅脱氧胆酸(CDCA),然后再分别与牛磺酸、甘氨酸结合形成结合型胆汁酸[7]。合成后的初级胆汁酸经胆盐输出泵(bile salt export pump,BSEP)存入胆囊,进食引起的胆囊收缩可以促进胆汁酸分泌进入肠道(图1)。肠道菌群中的拟杆菌(Bacteroides)、梭菌(Clostridium)、乳 酸杆菌(Lactobacillus)、双歧杆菌(Bifidobacterium)和李斯特菌(Listeria)产生的胆酸盐水解酶(BSH)可以使结合型胆汁酸发生去结合作用而形成游离胆汁酸;梭菌、梭杆菌(Fusobacterium)、消化球菌(Peptococcus)和假单胞菌(Pseudomonas)可以催化胆汁酸的脱硫作用;拟杆菌、真杆菌(Eubacterium)、梭菌、埃希氏菌(Escherichia)、埃格特菌(Eggerthella)、消化链球菌(Peptostreptococcus)和瘤胃球菌(Ruminococcus)可以将C3、C7和C12上的羟基脱去而形成次级胆汁酸,如脱氧胆酸(DCA)和石胆酸(LCA)[1,8]。
大约95%的初级胆汁酸和次级胆汁酸在肠道中都能被重吸收并经门静脉运回肝脏,但是次级胆汁酸中的石胆酸(LCA)基本上随粪便排出体外。肠-肝循环中损失的胆汁酸由肝细胞合成新的胆汁酸进行补足。胆汁酸重吸收后通过与回肠细胞的法尼醇X受体(farnesoid X receptor,FXR)和G蛋白偶联胆汁酸受体-5(G protein-coupled bile acid receptor-5,TGR-5)结合,促进肠细胞分泌成纤维细胞生长因子-19(FGF-19),后者再与成纤维细胞生长因子受体-4(fibroblast growth factor receptor-4,FGFR-4)结合,然后分别通过激活c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)和胞外信号调控激酶(extracellular signal-regulated kinase,ERK)两大信号通路降低胆固醇7α羟化酶(CYP7A1)的基因表达,从而负反馈性地抑制胆汁酸的合成[9]。
1.2 胆汁酸对肠道菌群结构的作用
肠道中的各种胆汁酸均可以通过直接或间接的方式抑制肠道菌群的生长[2],特别是当胆酸(CA)经过脱羟基作用转化成次级胆汁酸脱氧胆酸(DCA)后,其抑菌能力可增加10倍以上。低剂量的各种胆汁酸可以影响细菌细胞膜的流动性和通透性,而高剂量的胆汁酸可以直接与细菌细胞膜上的磷脂结合并破坏膜内蛋白质结构,导致细菌细胞内部的各种酶以及其他内容物流出,从而达到抑制肠道菌群过度生长的效果[4]。高脂饮食引起的胆汁酸分泌量增加和胆汁酸成分改变可以显著影响肠道菌群的种类和数量,而且不同的肠道菌群成员对不同的胆汁酸成分的敏感度也各不相同。
当给大鼠喂食胆酸后可引起厚壁菌门(Firmicutes)的丰度增加,而拟杆菌门(Bacteroidetes)的丰度减少,最终导致厚壁菌门与拟杆菌门之间的比例(F/B比值)增加。当给小鼠喂食乳源脂肪后会导致牛磺胆酸(TCA)的水平升高,而沃氏嗜胆菌(Bilophila wadsworthia)等条件致病菌的丰度也随之增加[10]。Janssen等[11]研究发现给小鼠喂食牛磺胆酸(TCA)不仅会引起肠道菌群结构改变,而且还可以加剧肝脏炎症反应和肝纤维化,表明胆汁酸代谢可以通过引起肝脏的免疫反应而抑制某些特定微生物生长,从而间接影响肠道菌群结构。胆汁酸与法尼醇X受体(FXR)结合后可以调节肠道中抗菌物质的基因表达,通过激活小肠的防御系统来防止细菌过度生长[12]。鹅脱氧胆酸(CDCA)与FXR受体结合后可以引起肠道上皮细胞分泌具有广谱抗菌活性的抗菌肽(如cathelicidin等),进一步发挥抗菌作用[4]。此外,高脂饮食引起的肝细胞脂肪堆积和胰岛素抵抗可导致胆汁酸淤积和胆汁酸成分改变,从而导致肠道菌群失衡,进一步加重胆汁酸代谢紊乱和肝脏炎症反应[13]。
2 胆汁酸和肠道菌群共同影响非酒精性脂肪性肝病的发生
2.1 胆汁酸代谢对非酒精性脂肪性肝病的影响
近年来,我国人群中非酒精性脂肪性肝病(NAFLD)的发病率快速上升,且发病年龄呈现越来越年轻化的趋势。研究表明,生命早期的营养环境可以影响胎儿长大后患上非酒精性脂肪性肝病的风险,特别是母体孕期营养异常对胎儿的胆汁酸代谢、线粒体功能和巨噬细胞活化的不良影响均可以增加胎儿成年后对非酒精性脂肪性肝病的易感性[14]。胆汁酸代谢异常引起的脂代谢异常和肝细胞脂肪堆积是引起非酒精性脂肪性肝病发病的重要因素[15]。“二次打击”学说(the“two-hit”theory)认为,外周脂肪分解增加和胰岛素抵抗(insulin resistance,IR)构成的“初次打击”会引起肝细胞脂肪酸合成积累,并对肝细胞产生多种损伤后果;而肝细胞脂质过氧化、氧化应激和内毒素入血等多种损伤后果作为“二次打击”可进一步引起肝细胞发生气球样病变和坏死性炎症,并逐渐发展为肝纤维化和肝硬化,最后甚至会导致肝细胞癌(HCC)。因此,胆汁酸代谢异常和胰岛素抵抗引起的肝脏脂肪变性和炎症感染是导致非酒精性脂肪性肝病的重要影响因素[16]。

图1 胆汁酸代谢和肠道菌群的对话交流机制Fig.1 The crosstalk between bile acid metabolism and intestinal microbiota
胆汁酸的合成和转运调节对于维持肠-肝循环中的胆汁酸平衡和稳定具有重要意义。胆汁酸淤积造成的肝细胞损伤可以引起肝脏炎症反应,并最终可导致非酒精性脂肪性肝病的发生。不同种类胆汁酸的疏水性不同,疏水性最强的胆汁酸是石胆酸(LCA),其次分别是脱氧胆酸(DCA)、鹅脱氧胆酸(CDCA)、胆酸(CA)和熊去脱氧胆酸(UDCA),一般认为疏水性越强的胆汁酸其相应的细胞毒性就越大。由于次级胆汁酸的疏水性总体上大于初级胆汁酸,因此脱氧胆酸(DCA)和石胆酸(LCA)不仅可以诱导细胞损伤,还具有一定的致癌性[17]。由肠道菌群失衡引起的疏水性胆汁酸 (如DCA和CDCA)在肝脏内的积聚可导致胆汁淤积性肝损伤,疏水性胆汁酸浓度升高后会破坏肝细胞膜,激活磷脂酶A2(phospholipase A2,PLA2),并引起肝细胞线粒体损伤,随后通过增加活性氧(reactive oxygen species,ROS)水平激活RAS信号通路,最终可导致肝细胞癌(HCC)的发生。然而亲水性较强的熊去氧胆酸(UDCA)及其与牛磺酸结合形成的衍生物牛磺熊去脱氧胆酸(TUDCA)则被证实具有细胞保护作用和显著的抗炎效果[18]。胆汁酸通过与FXR受体和TGR-5受体结合不仅可以调节胆汁酸的合成和转运,而且还参与调控机体能量代谢、肝脏再生和炎症反应等生理过程[19]。Trabelsi等[20]研究发现胆汁酸与FXR/TGR-5受体结合后可增加胰高血糖素样肽(glucagon-like peptide-1,GLP-1)的分泌,并可通过刺激胰岛β细胞分泌胰岛素调节糖代谢。
2.2 肠道菌群对非酒精性脂肪性肝病的影响
慢性肝病患者体内的胆汁酸代谢异常通常也会引起肠道菌群结构的紊乱,其中肠道内胆汁酸谱的变化可导致肠道菌群物种的多样性和丰度均发生改变,例如:胆汁酸淤积可引起小肠细菌过度生长(small intestinal bacterial overgrowth,SIBO),从而严重影响小肠的消化和吸收功能。研究发现,非酒精性脂肪性肝病患者的肥胖和胰岛素抵抗会引起双歧杆菌(Bifidobacterium)和艾克曼菌(Akkermansia muciniphila)等益生菌的丰度明显下降;同时,丁酸弧菌(Butyrivibrio crossotus)、粪栖杆菌(Faecalibacterium prausnitzii)、罗斯氏菌(Roseburia inulinivorans)和厌氧棍状菌(Anaerotruncus colihominis)等丁酸盐产生菌的丰度也显著降低[21]。这些肠道益生菌的数量减少及其产生的短链脂肪酸的减少会导致肠道屏障功能受损,进而引起肠道通透性增加,因此更容易发生肠道细菌易位和有毒代谢产物进入血液。另外,非酒精性脂肪性肝病患者肠道中的条件致病菌,如瘤胃球菌(Ruminococcus gnavus)、普雷沃氏菌(Prevotella copri)、弯曲杆菌(Campylobacter)和志贺氏菌(Shigella)等,则出现了增长趋势。结构失衡的肠道菌群会产生大量的有毒代谢产物,如脂多糖、内生性乙醇和三甲胺等,这些小分子化合物经血液循环进入肝脏后会进一步加重肝细胞损伤。
3 胆汁酸和肠道菌群影响非酒精性脂肪性肝病的作用机理
3.1 胆汁酸代谢影响非酒精性脂肪性肝病的作用机理
胆汁酸通过与FXR受体和TGR-5受体结合调控机体的免疫稳态和炎症应答,进一步影响肝脏细胞脂肪变性、细胞损伤和细胞凋亡等生理过程[22]。FXR受体是一种可以在多种组织和器官中表达的核受体,在肝脏细胞和回肠细胞中的表达量最高,在肾脏、心脏、胸腺、脾脏、卵巢和睾丸细胞中也可以表达[7]。FXR受体能够与启动子区域结合并启动多种靶基因表达,从而调节炎症反应、胆汁酸代谢和糖脂代谢等多种生理过程(图2)。FXR受体被胆汁酸激活后能够通过与胱冬肽酶-1(caspase-1)相互作用抑制NLRP3炎性小体的激活,并可通过减少白细胞介素-1β(interleukin-1β,IL-1β)等炎症因子的释放减轻非酒精性脂肪性肝病的症状[23]。FXR基因敲除的小鼠肝脏会出现次级胆汁酸比例增加以及淋巴细胞和中性粒细胞浸润现象,而FXR基因过表达则可以缓解炎症感染造成的肝损伤[24]。不同胆汁酸与FXR受体的亲和力不同,其中鹅脱氧胆酸(CDCA)与FXR受体的结合能力最强,然后依次是胆酸(CA)、脱氧胆酸(DCA)和石胆酸(LCA),但是UDCA不仅不能激活FXR受体,反而起到抑制FXR受体的作用。因此,通过干预初级胆汁酸和次级胆汁酸的比例来激活FXR受体可以作为治疗非酒精性脂肪性肝病的新策略[25]。
TGR-5受体是一种可以被胆汁酸激活的G蛋白偶联受体(G protein-coupled receptor,GPCR),在胆囊、胎盘、肺、脾、肠道中表达,也可以在棕色脂肪组织、白色脂肪组织、骨骼肌和骨髓中表达[6~7]。当TGR-5受体被次级胆汁酸(如LCA和DCA)激活后,可通过激活环磷腺苷(cyclic adenosine monophosphate,cAMP)/蛋白激酶 A(protein kinase A,PKA)信号途径引起NLRP3炎性小体的单个氨基酸(Ser291)发生磷酸化,从而发挥抗炎作用[26]。虽然TGR-5基因敲除小鼠具备常规表型和繁殖能力,但是胆汁酸池却明显减少,表明TGR-5受体对维持胆汁酸稳态具有重要作用。因此,调节胆汁酸代谢和TGR-5受体信号通路也可以作为治疗非酒精性脂肪性肝病的潜在策略[27]。
3.2 肠道菌群影响非酒精性脂肪性肝病的作用机理
肠道屏障功能对于维持肠道菌群和宿主免疫系统之间的平衡有重要作用,肠道菌群通过生成一系列代谢产物(如短链脂肪酸、乙醇和三甲胺等)以及相关的信号通路对非酒精性脂肪性肝病的发生和发展产生重要的影响[28]。通常情况下,肠道菌群产生的短链脂肪酸(SCFAs)激活G蛋白偶联受体(GPR41和GPR43)后可以抑制肠道蠕动并加强营养吸收和能量获取,从而提高肝脏脂肪生成,促进非酒精性脂肪性肝病的发生和发展。最新研究发现,丁酸盐可以通过激活过氧化物酶体增殖物激活受体α(peroxisome proliferators-activated receptors α,PPARα)和抑制 NLRP3炎症信号通路来降低氧化应激和炎症反应,从而起到缓解非酒精性脂肪性肝病的作用[29]。肠道微生物产生的内源性乙醇可以促进炎症因子的释放,并且内源性乙醇代谢生成的乙醛可以通过影响紧密连接蛋白的功能而增加肠道渗透性。另外,肠道菌群可以通过代谢胆碱生成三甲胺(trimethylamine,TMA)和氧化三甲胺(trimetlylamine oxide,TMAO),影响葡萄糖代谢并增加胰岛素抵抗;同时,还可以通过增加血清中趋化因子(chemokines)的水平促进非酒精性脂肪性肝病的发展[30]。
条件致病菌产生的病原体相关分子模式(pathogen-associated molecular patterns,PAMPs)可与肝脏库普弗细胞(Kupffer cell)表面的模式识别受体(pattern recognition receptor,PRR)结合,然后激活 Toll样受体-4(toll-like receptor-4,TLR-4)/髓样分化因子-88(myeloid differentiation factor-88,MyD-88)和核因子 κB(nuclear factor-κB,NF-κB)等炎症信号通路,引起肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)、IL-1β 和 IL-6 等促炎细胞因子的释放,从而促进非酒精性脂肪性肝病的发生和发展[31]。胆汁酸(尤其是牛磺酸和甘氨酸结合型的胆汁酸)可通过激活巨噬细胞M1和M2表面的TGR-5受体分别起到促炎和抗炎的作用,而肠道菌群则可通过产生胆盐水解酶(BSH)解离结合型的胆汁酸,从而改变巨噬细胞M1和M2分泌的促炎细胞因子和抗炎细胞因子的水平,最终影响肝脏的免疫平衡状态。当肠道菌群产生的脱氧胆酸(DCA)和石胆酸(LCA)等次级胆汁酸过少时,将会导致FXR受体活性降低,进一步加剧机体的炎症状态;反之,如果肠道菌群产生的次级胆汁酸(尤其是DCA)过多,大量具有细胞毒性的次级胆汁酸则会通过产生活性氧(ROS)造成细胞DNA损伤,导致肝细胞癌的发生[32]。此外,肠道菌群通过调节胆汁酸代谢可进一步激活腺苷酸活化的蛋白激酶(AMP-activated protein kinase,AMPK)/磷酸化乙酰辅酶A羧化酶(acetyl-CoA carboxylase,ACC)和胆固醇调节元件结合蛋白-1c(sterol regulatory element-binding protein-1c,SREBP-1c)/硬脂酰辅酶A去饱和酶-1(stearoyl-CoA desaturase-1,SCD-1)等氧化应激和脂代谢相关信号通路,从而影响非酒精性脂肪性肝病的发生和发展[33]。
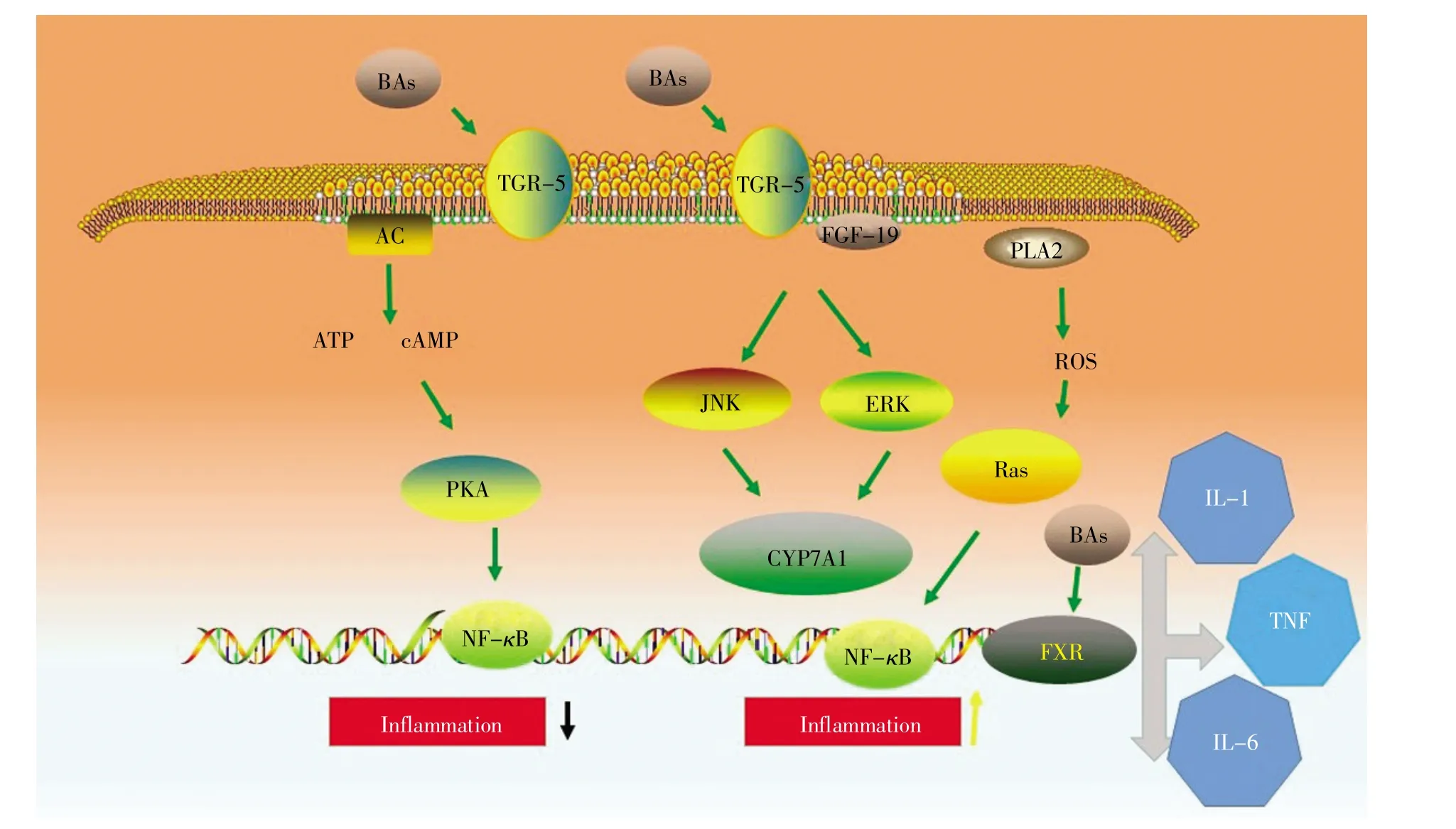
图2 胆汁酸代谢和肠道菌群影响非酒精性脂肪性肝病的作用机理Fig.2 Influences on the development of NAFLD by bile acid metabolism and intestinal microbiota
4 总结与展望
非酒精性脂肪性肝病是全球流行性的代谢性肝损伤,与生活方式和综合代谢紊乱等因素都有密切关系。通常情况下,肝脏被认为是最重要的分解肠道毒素的器官,大约70%的肝脏血液供给都来自于肠门静脉,因此肠道菌群产生的有毒代谢产物(如乙醇、乙醛和氨等物质)基本都在肝脏中被分解代谢。此外,肠道微生物还可以调节肝脏库普弗细胞的活性和炎症相关的细胞因子的产生,从而影响肝脏的免疫稳态和炎症反应[34]。近年来,随着宏基因组学、宏转录组学和代谢组学等多组学技术手段的发展与进步,越来越多的研究发现胆汁酸代谢异常和肠道菌群紊乱共同导致的肝细胞内胆汁酸淤积和肝脏脂肪堆积对于推动非酒精性脂肪性肝病的发生和发展有重要影响[35]。
目前,临床上主要通过调控代谢水平和控制生活方式来治疗非酒精性脂肪性肝病,但是治疗效果十分有限。最新研究表明,通过调节肠道菌群结构和胆汁酸组成来治疗非酒精性脂肪性肝病已经成为重要的研究方向[36]。近年来,人们通过探索传统中草药对非酒精性脂肪性肝病患者的肠道菌群结构和胆汁酸组成的改善作用,发现将中草药修复肠道屏障功能、改善机体免疫稳态等生物学功能与抗氧化、抗炎等治疗优势密切结合,可以起到较好的治疗效果[37]。此外,利用粪菌移植(fecal microbiota transplantation,FMT)的方法重建肠道菌群,可进一步降低内毒素、内生性乙醇等代谢产物向肝脏的转运,并减少肝细胞膨胀和炎症反应,因此FMT被证明是预防和治疗非酒精性脂肪性肝病的有效手段[38]。
综上所述,通过研究非酒精性脂肪性肝病患者的肠道菌群和胆汁酸之间的对话交流机制,可以将调节免疫稳态与改善代谢水平相结合,从而为治疗非酒精性脂肪性肝病提供新靶点。
猜你喜欢
肝博士(2024年1期)2024-03-12 08:38:22
现代临床医学(2022年4期)2022-09-29 07:38:08
肝博士(2022年3期)2022-06-30 02:49:06
理化检验-化学分册(2021年10期)2021-11-29 14:50:42
肝博士(2021年1期)2021-03-29 02:32:02
肝博士(2020年5期)2021-01-18 02:50:26
中西医结合肝病杂志(2020年2期)2020-10-27 02:18:42
中国卫生标准管理(2015年16期)2016-01-20 09:26:29
中国当代医药(2015年9期)2015-03-01 02:02:12
西南军医(2015年2期)2015-01-22 09:09:27
