
Recurrent acute liver failure associated with novel SCYL1 mutation:A case report
2019-04-16 08:23:34JiaQiLiJingYuGongKniselyMeiHongZhangJianSheWang
World Journal of Clinical Cases 2019年4期
Jia-Qi Li,Jing-Yu Gong,A S Knisely,Mei-Hong Zhang,Jian-She Wang
Abstract
Key words: SCYL1;Recurrent acute liver failure;Whole-exome sequencing;Case report
INTRODUCTION
An unusual form of acute liver failure(ALF)occurs in children.Starting in infancy,episodes of hepatic insufficiency recur.These episodes may be preceded by fever(and thus be ascribed to antipyretics,in particular paracetamol)[1-3].They may not be restricted to childhood[4,5]and may be fatal[6].They are characterized by clinicallaboratory findings that include marked elevations in serum transaminase activity,severe coagulopathy,conjugated hyperbilirubinemia,and normal or only slightly elevated serum gamma-glutamyl transpeptidase activity[2,4].Hepatic crises,with less severe injury,also are seen[1-5].The causes of recurrent ALF(RALF),as this disorder is termed,are incompletely understood,although instances have been ascribed to biallelic mutation inSCYL1(MIM:607982)[7]orNBAS(MIM:608025)[4].NBASdisease is multisystem in some patients[3,6,8]and manifest only as RALF in others[2,4].The few patients yet reported withSCYL1disease have exhibited isolated RALF in conjunction with a range of mild to more marked neurological phenotypes,including spinocerebellar ataxia and neurodegeneration[7,9-11].In one,recurrent ventilatory failure was clinically prominent;RALF was not described[12].
We report a novel homozygous mutation inSCYL1in a Han Chinese boy who had RALF with onset in infancy and in whom,aged 8 years 6 mo,mild neurologic dysfunction and bilateral femoral head abnormalities were identified.Our observations expand the clinical and mutational spectrum ofSCYL1disease.
CASE PRESENTATION
Chief complaints
A 7-year-old boy was admitted to our hospital due to recurrent episodes of ALF.
History of present illness
The patient was well till age 14 mo,when a fever was treated with the non-steroidal anti-inflammatory agent nimesulide.Jaundice developed,with elevated transaminase activity.Injury fell short of frank ALF,constituting instead a “liver crisis”.Two episodes of ALF followed at ages of 3 years 6 mo and 6 years 3 mo(Table1).Clinical,laboratory,and imaging-study findings between episodes were normal.At the age of 7 years,shortly after his third RALF episode,he was referred to our hospital for etiological diagnosis.
History of past illness
His parents and he denied any history of disease beyond that summarized above.
Personal and family history
He was born at term(weight 3 kg)by elective cesarean section after an uncomplicated pregnancy.He is the second child of a Han Chinese couple who deny consanguinity.His sibling,a brother,is well.
Physical examination upon admission
Physical examination on admission found no abnormalities.
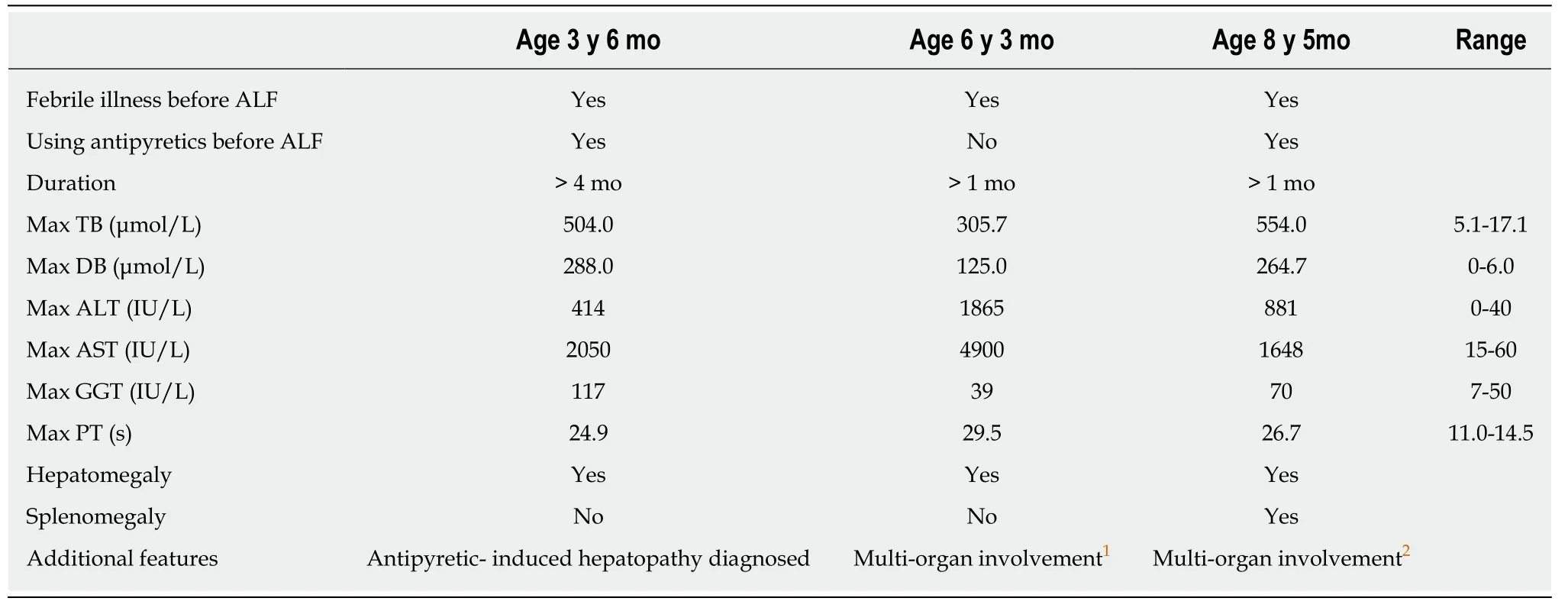
Table1 Clinical phenotype in three fever-associated acute liver failure episodes
Laboratory examinations
Blood,urine,and stool routine test results,viral serological markers(hepatotropic viruses,Epstein-Barr virus,cytomegalovirus,and human immunodeficiency virus 1),biomarkers of toxoplasma infection,of thyroid function,and of hepatobiliary injury,and values for cholesterol,triglycerides,creatine phosphokinase,glucose,ceruloplasmin,blood lactate,blood ammonia,carbonyldiamide,creatinine,uric acid,urine organic acids,alpha-fetoprotein,and immunoglobulin levels were unremarkable.The value of 25-(OH)D3was 13.69 ng/mL(normal:30-100 ng/mL).
Imaging examinations
Cranial magnetic resonance imaging showed a cyst(90 mm × 30 mm)in the cisterna magna and a relatively small cerebellum.The optic nerves were normal.Abdominal ultrasonography and echocardiography showed no significant abnormalities.
Other examinations
Findings on ophthalmologic evaluation were unremarkable.Liver biopsy found mildly abnormal architecture,without fibrosis,in a pattern suggesting disordered perfusion;no ultrastructural abnormalities were apparent.The Denver developmental screening test,which covers gross motor,language,fine motor-adaptive,and personal-social skills[13],found a low developmental quotient(< 47,normal > 70)and mental index(63,normal > 70).
Whole-exome sequencing
With the approval of the ethics committee of the Jinshan Hospital of Fudan University(Jinyi Lunli Keyan-2014-13-01)and after obtaining written informed consent from the parents of the patient,DNA was isolated from peripheral blood samples obtained from the proband,his brother,and his parents.Whole-exome sequencing was conducted as described[2](Genesky Biotechnologies,Shanghai,China).Exomes were captured using a SureSelectXT Human All Exon V6 kit(Agilent Technologies,Foster,CA).Sequencing(250 bp paired-end reads)was performed using the Illumina hiseq2500 platform following the manufacturer's instructions(Illumina,San Diego,CA).Burrows-Wheeler Aligner software(http://bio-bwa.sourceforge.net/)was used for read alignment(hg19;NCBI build 37;February,2009).Variant calling was performed with Varscan(http://varscan.sourceforge.net/)and GATK Haplotype-Caller(https://software.broadinstitute.org/gatk/best-practices/).Mean coverage of the exome was 140× with 91% of the exome covered at least 10 times and 80% covered at greater than 20×.The proband's variants were filtered for minor allele frequency <1% against Thousand Genomes Project(http://www.1000genomes.org/home),NHLBI Exome Sequencing Project(http://evs.gs.washington.edu/EVS/),Exome Aggregation Consortium Server(http://exac.broadinstitute.org/),and Genesky inhouse databases.Variants in ALF-associated genes(Supplementary file 1)assessed by SIFT(http://sift.jcvi.org/),Polyphen-2(http://genetics.bwh.harvard.edu/pph2/),or MutationTaster[14](http://www.mutationtaster.org/)as pathogenic and present in accord with inheritance modes were considered of interest.Sanger sequencing was used to confirm variants in the proband and to seek them in his parents and brother.
Identification of a novel homozygous mutation in exon 1 in SCYL1
The homozygous mutation c.92_93insGGGCCCT,p.(H32Gfs*20)in exon 1 inSCYL1(NM_020680)was identified in the proband.His brother did not harbor this mutation.Each of their parents was heterozygous for the mutation,consistent with recessive inheritance.The mutation detected by exome sequencing was again identified on Sanger sequencing(Supplementary file 2).No other variant of interest was identified in any ALF-associated gene.The insertion of GGGCCCT between nucleotides 92 and 93 within exon 1 is predicted to result in the substitution of 19 abnormal amino acid residues(His32-Val50),followed by a stop codon at codon 51.MutationTaster predicts that this results in nonsense-mediated mRNA decay[14].The variant is novel;it was not recorded in Exome Aggregation Consortium Server,NHLBI Exome Sequencing Project,Thousand Genomes Project,or Genesky in-house databases.
FINAL DIAGNOSIS
SCYL1disease.
TREATMENT
Supportive care during episodes of ALF or crises followed accepted guidelines and was not specific for any particular form of hepatic dysfunction.They included antibiotics to resist bacterial infection,”liver protectant drugs”(D-glucurono-3,6-lactone,diammonium glycyrrhizinate,polyene phosphatidylcholine,etc.),cholagogues and choleretics(ursodeoxycholic acid,cholestyramine,etc.),prednisone,and supplementary fat-soluble vitamins A,D,E,and K.
参议员们开了个特别会议,派出一个代表团对她进行了访问。他们敲敲门,自从八年或者十年前她停止开授瓷器彩绘课以来,谁也没有从这大门出入过。那个上了年纪的黑人男仆把他们接待进阴暗的门厅,从那里再由楼梯上去,光线就更暗了。一股尘封的气味扑鼻而来,空气阴湿而又不透气,这屋子长久没有人住了。黑人领他们到客厅里,里面摆设的笨重家具全都包着皮套子。黑人打开了一扇百叶窗,这时,便更可看出皮套子己经坼裂;等他们坐了下来,大腿两边就有一阵灰尘冉冉上升,尘粒在那一缕阳光中缓缓旋转。壁炉前已经失去金色光泽的画架上面放着爱米丽父亲的炭笔画像。
OUTCOME AND FOLLOW-UP
The patient had his fourth episode of RALF at 8 years 5 mo(Table1).During resolution of this episode,liver biopsy was conducted again.Alanine transaminase and aspartate aminotransferase values 2 d before liver biopsy were 91 IU/L(expected:0-40 IU/L)and 476 IU/L(normal:15-60 IU/L).Ballooning change of hepatocytes,some with cytoplasmic bile pigment,and hepatic fibrosis were observed.A comprehensive skeletal survey by radionuclide bone scan showed bilateral hip joint widening and bilateral femoral head flattening highly suggestive of bilateral femoral head necrosis.The patient failed finger-to-nose testing and Romberg testing,indicating cerebellar ataxia.However,muscle strength,muscular tension,stretch reflexes,deep sensory perception,and motor function were normal.At last evaluation,aged 8 years 7 mo,biomarker values in the proband were normal.
DISCUSSION
SCYL1encodes a kinase-like protein in the SCY1-like family[15,16].SCYL1 may function as a scaffolding protein,involved in Golgi-to-endoplasmic reticulum retrograde transport and in Golgi integrity[17-19].SCYL1 also acts in nucleocytoplasmic shuttling of tRNAs[20].Biallelic mutations inSCYL1were first identified in 2015 as an important cause of a hepatocerebellar neuropathy syndrome(spinocerebellar ataxia,autosomal recessive 21;MIM:616719)[7].
To date,only 5 reports have described 14 patients harboring homozygous or compound heterozygous mutations inSCYL1[7,9-12].Thirteen patients had similar hepatic features[7,9-11];in the 14th,features of liver disease were present,but recurrent ventilatory failure dominated clinical illness[12].Neurologic phenotypes varied,including peripheral neuropathy,ataxia,tremor,stuttering,speech-development delay,intellectual disability,microcephaly,and abnormal intracranial magneticresonance imaging findings.Six SCYL1-deficient patients had cerebellar atrophy and 2 siblings had optic nerve thinning[7,10,12].
Our patient had a classic hepatic phenotype,with one episode of liver crisis(aged 14 mo)and 3 episodes of acute liver failure(aged 3 years 6 mo,6 years 3 mo,and 8 years 5 mo).Each episode was preceded by a febrile illness.Hepatobiliary-injury biomarker values recovered completely between episodes.Not all episodes of febrile illness led to hepatic crisis.Liver biopsy conducted after the third RALF episode(7 years)and during resolution of the fourth RALF episode(8 years 6 mo)revealed slightly abnormal architecture and hepatic fibrosis,respectively.
In our patient,relative cerebellar hypoplasia may be secondary to the cyst in the cisterna magna;vermis atrophy was not identified,and optic nerves were not thinned.Clinical neurologic features are at this writing minimal,with low developmental quotient and mental index,minor intracranial abnormalities,and abnormal coordination tests.These may worsen as the patient ages,or other abnormalities may yet declare themselves.Neurologic abnormalities usually are noted inSCYL1disease after liver disease has appeared(Supplementary file 3).To evaluateSCLY1in infants or children with RALF thus is indicated even if neurologic disease is inapparent.Long-term follow-up is necessary for best correlation between mutation and phenotypic consequences.
Worth noting is that skeletal abnormalities were observed in 8 of 14SCYL1-deficiency patients.Skeletal phenotypes varied,including short stature,hip dysplasia,coronal clefting of ribs,scoliosis,lumbar lordosis,and thoracic vertebral abnormalities[9-11].Our patient had bilateral femoral head disease,not reported before in association withSCYL1mutation.At least some of the skeletal abnormalities described appear primary rather than resulting from neuromuscular dysfunction,suggesting that such abnormalities may be an important clinical manifestation ofSCYL1disease.Systemic skeletal examinations thus may be essential for children withSCYL1deficiency.
Early identification of the cause of ALF is fundamental for disease-specific management.In our patient,assignment of diagnosis to mutation in a specific gene shifted the context in which we viewed his findings on examination,changing our expectations for the course of what now can be considered as a multisystem disorder.For instance,during the fourth RALF episode,after genetic diagnosis,a comprehensive skeletal survey and more detailed neurologic examination were performed.Our care has become better focused and,with the progress of the disease,may perhaps permit orthopedic,physiatric,and neurologic intervention for directed support.In addition,genetic diagnosis can also help in prenatal screening.
Our patient is the first East Asian described withSCYL1-associated RALF.TheSCYL1mutation c.92_93insGGGCCCT,p.(H32Gfs*20),which he harbors in homozygous state,is predicted to result in nonsense-mediated decay of a prematurely terminated RNA transcript.Although we lack direct evidence for lack of SCYL1 expression in our patient,we thus consider him the 15thidentified patient with RALF due toSCYL1disease.We call attention to his clinical phenotype,with scant neurologic dysfunction identified thus far and relatively late-onset skeletal disease,although our findings are -without life-long follow-up -necessarily incomplete.We suggest that even if neurologic features of disease are few(“atypical”SCYL1disease),SCYL1defects should be considered as possibly underlying early-onset fever-related RALF.We also note as deserving further study the association between skeletal abnormalities andSCYL1disease.
CONCLUSION
Even if neurologic features of disease are few,SCYL1 defects should be considered as possibly underlying early-onset fever-related RALF.Systemic skeletal examinations are in order for children withSCYL1deficiency.
ACKNOWLEDGEMENTS
The authors are grateful for the support of the patient's family and thank referring physicians,nurses,and technical staff.
猜你喜欢
工程科学学报(2021年11期)2021-11-17 08:54:50
品牌研究(2020年4期)2020-12-20 11:04:51
艺术大观(2019年31期)2019-10-12 05:37:45
疯狂英语·新读写(2019年1期)2019-01-16 03:52:44
课外生活(小学1-3年级)(2017年5期)2017-06-10 20:08:37
小樱桃·童年阅读(2016年5期)2016-05-25 02:32:41
剑南文学(2015年14期)2015-11-23 00:47:43
幼儿教育(2015年32期)2015-02-14 05:45:40
中华女子学院学报(2012年6期)2012-03-25 13:52:28
太原城市职业技术学院学报(2010年12期)2010-08-15 00:49:04
 World Journal of Clinical Cases2019年4期
World Journal of Clinical Cases2019年4期
- World Journal of Clinical Cases的其它文章
- Rhombencephalitis caused by Listeria monocytogenes with hydrocephalus and intracranial hemorrhage:A case report and review of the literature
- Anterior cervical corpectomy decompression and fusion for cervical kyphosis in a girl with Ehlers-Danlos syndrome:A case report
- Primary hepatic leiomyosarcoma successfully treated by transcatheter arterial chemoembolization:A case report
- Long-term follow-up of a patient with venlafaxine-induced diurnal bruxism treated with an occlusal splint:A case report
- Hydrochloric acid enhanced radiofrequency ablation for treatment of large hepatocellular carcinoma in the caudate lobe:Report of three cases
- Therapeutic plasma exchange and continuous renal replacement therapy for severe hyperthyroidism and multi-organ failure:A case report