
表现为前哨脱髓鞘的原发性中枢神经系统淋巴瘤1例报告☆
2019-04-08 08:09:14马改英张令霞韩旭
中国神经精神疾病杂志 2019年2期
马改英 张令霞 韩旭
徐瑞雪* 陈丽萍 *○☆
原发性中枢神经系统淋巴瘤 (primary central nervous system lymphoma,PCNSL)是指发生在脑、脑膜、脊髓或眼的侵袭性非霍奇金淋巴瘤,为神经系统较少见的恶性肿瘤之一[1-2]。其发病机制尚不清楚。绝大多数的PCNSL在确诊前的数月或数年常表现为多灶性髓鞘脱失,即 “前哨脱髓鞘”,与特发性中枢神经系统炎性脱髓鞘不易鉴别。现报告我科收治的一例经病理证实为PCNSL的病例。
1 病例资料
患者女,43岁,2012年12月10日因突然出现双下肢麻木7 d就诊于当地医院。头颅MRI检查显示双侧额顶叶皮层下及双侧半卵圆中心多发T1WI等信号、T2WI和FLAIR高信号,考虑脱髓鞘。给予激素冲击治疗后好转,具体用药剂量和疗程等信息不详。2013年3月6日,该患者因四肢麻木无力伴左眼视力下降7天来我院就诊,查体:神清,言语不清。双上肢肌力5级,双下肢肌力5-级,双侧腱反射(+++),双侧 Babinski "s征(+),余未见明显异常。 行视觉诱发电位、头颅和胸椎MRI检查。视觉诱发电位提示双眼P100潜伏期延长。MRI结果提示,脑内T1WI等信号、T2WI和FLAIR高信号病灶范围较前增大,且新增双侧深部白质,双侧锥体束走行区、双侧小脑半球、双侧桥臂T1WI等信号、T2WI和 FLAIR高信号病灶(见图 1B),同时胸髓内亦见多发 T1WI等信号、T2WI高信号病灶 (见图1A),考虑多发性硬化(multiple sclerosis MS)。给予激素冲击治疗500 mg×5 d后依次减半,症状好转,15 d以后出院,嘱患者规律口服小剂量激素。2013年6月8日,该患者因四肢无力、视物不清3 d,加重1 d再次入院。查体:神清,语言含糊,左上肢肌力5级,右上肢肌力3级,左下肢肌力3级,右下肢肌力3级,四肢肌张力正常,双上肢腱反射(+++),双下肢腱反射(++++),双侧 Babinski "s征(+),双侧踝阵挛(+),右下肢及右腹部感觉减退,共济查体欠合作。头颅MRI提示脑内多发白质脱髓鞘,左顶叶新增占位性病变,增强后周边明显强化(见图1C)。2013年6月14日行开颅手术,术后病理提示左顶叶皮质下区弥漫性大B细胞淋巴瘤,HE染色下弥漫性异常增值的B细胞 (见图2A)免疫组化结果显示 CD45(+++)、CD45R0(+~++)、CD20(++~+++)(见图 2B)、Bcl-2(-)、Bcl-6(+)、CD5(+~++)、Ki-67(80%)、GFAP(-)、S-100(-)、CD34(-)。 该患者术后13 d出院,3周后于外院放疗,术后17个月去世。
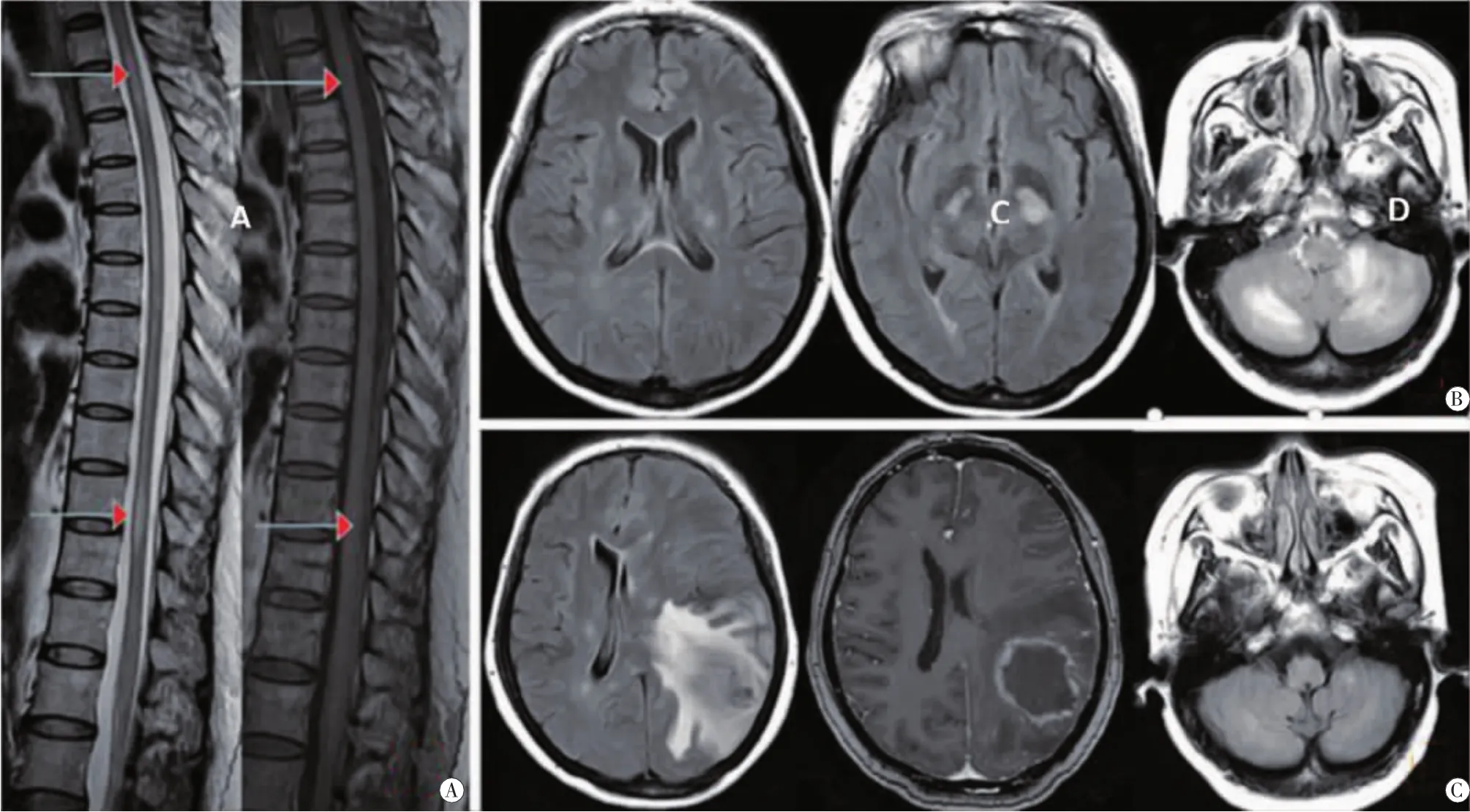
图1 头颅和胸椎MRIA胸椎MRI(2013年03月09日):矢状位胸髓内多发斑片状长 T2等T1信号(红色箭头所示);B头颅MRI(2013年03月09日):颅内多发高FLAIR信号 (长T2、等T1信号);C头颅MRI(2013年06月09日):双侧侧脑室体旁白质多发多发斑点状高FLAIR信号,小脑半球病灶较前变小,新增左顶叶占位伴周围大面积水肿,增强扫描周边可见明显强化
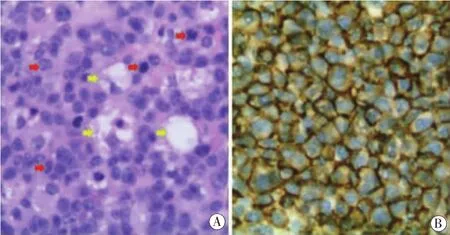
图2 组织病理学(2013年06月14日):A:HE染色:弥漫异常增值的B细胞(红色箭头)和少量巨噬细胞(黄色箭头,400×);B:细胞免疫染色: CD20(++~+++)(400×)
2 讨论
该患者在病理确诊为PCNSL之前,共有3次临床发作,临床症状与责任病灶对应,且持续24 h以上,任意2次临床发作间隔1个月以上。影像学上不同位置多发的T2病灶第二次较第一数目增多,范围也较前变大,满足MRI时间、空间多发。患者病情反复发作且经激素冲击治疗后症状明显改善。符合MS 2010年McDonald的诊断标准[3]。但当临床症状第三次突然发作,左顶叶新增肿瘤病变时,我们便不得不审视之前的诊断是否正确,多发性硬化和PCNSL发生在同一患者非常罕见,可查到的文献中只有一例一位20岁女性间隔8年后诊断为PCNSL,提示该患者同时患有多发性硬化和PCNSL。ALDERSON等[4]报告4例年龄在49岁至58岁之间的健康、免疫功能良好的患者,初步活检显示两例脱髓鞘,一例为非特异性炎症,一例为正常大脑。7~11个月后,每位患者在大脑不同区域出现新的症状性病变,活检显示为B细胞PCNSL。他们认为首次活检结果可能代表宿主对肿瘤的免疫。当局灶性实质病变显示非特异性或脱髓鞘性组织病理学改变时,应考虑PCNSL前哨病变。之后也有文献报道[4-5]的部分PCNSL患者在确诊前数月至数年可存在自发缓解或对激素敏感的“前哨炎性脱髓鞘病变”。1996年 ALDERSON等[3]首次将PCNSL之前的炎性脱髓鞘命名为“前哨病灶”,病理学上,“前哨病灶”以T淋巴细胞、巨噬细胞浸润为主,罕见B淋巴细胞[4-5]。关于其发生机制提出两种假说[4]:①前哨病灶是机体针对淋巴瘤的早期免疫反应,淋巴瘤可能因宿主免疫而处于休眠状态;②肿瘤细胞克隆时,其免疫球蛋白基因不断发生突变,前哨病灶是逃脱肿瘤的破坏表现。所以回顾该病例,虽然很遗憾早期无病理结果证实病变性质,但我们认为早期影像学表现的及临床过程与文献报道病例均相符,所以PCNSL从一开始就可能是肿瘤潜伏期多发的脱髓鞘病变,而不是MS合并PCNSL。
本例患者在PCNSL确诊前持续应用激素治疗,肿瘤的形成是否与激素诱导有关,HUSSEINI等研究[6]报告激素使用前行活检,病理证实“前哨病变”为炎症性破坏和局灶性脱髓鞘,后因占位效应明显再次行活检,病理证实是PCNSL,两次活检前均未激素治疗。BARRANTES等[7]尸检证实表现为前哨脱髓鞘的PCNSL患者活检前仅有短时间小剂量激素用药史。LEBRUN和他的同事对法国7418名多发性硬化症患者进行了详尽的多中心评估,评估疾病修饰疗法和癌症发病率的影响,他们没有发现在诊断出多发性硬化症后癌症风险增加的证据[8]。目前尚缺乏激素诱导PCNSL的证据。
目前PCNSL确诊依靠病理组织活检。文献[6,9-12]报告,组织病理学上确诊的弥漫性大B细胞淋巴瘤最初表现为脱髓鞘病变,炎症明显,没有淋巴瘤细胞的证据,很难与炎症性脱髓鞘疾病,特别是与多发性硬化相鉴别[3,13],BARRANTES等[7]比较了8例类固醇治疗后诊断为PCNSL和9例明确诊断为MS患者的脑组织病理特点,PCNSL患者组织学分析显示髓鞘脱失呈局灶性斑片状表现,轴索(神经丝蛋白免疫染色)和髓鞘(Luxol法快速蓝染)显著减少,浸润 T淋巴细胞数目是MS的5倍。免疫组化提示,肿瘤细胞表达 B细胞标记物,如 CD20、CD79a、CD19、CD22和 PXA5[14],其他标记物还包括 BCL6(60%~80%),MUM1/IRF4 (90%),CD10(10%~20%)和 BCL2[15]。 与 PCNSL 相比,MS髓鞘大量脱失、轴索部分保留,轴索肿胀变形、大量反应性星形细胞生成、血管周围大量淋巴细胞呈“套袖”样浸润、大量吞噬细胞生成、大量髓鞘再生。免疫组化显示,MS 患者的 GFAP、Olog-2,S100、NF、LCA、CD3、CD34、CD68及P53染色阳性率均达100%[16]。
KVARTA等[13]回顾性分析16例病理证实为PCNSL的个案报道,结果发现该疾病具有如下特点:①脑白质有前哨病灶;②好发于中老年女性;③既往无临床事件或影像学提示MS;④病情迅速恶化;⑤类固醇依赖;⑥影像学表现为随着时间的推移颅内病灶或强化病灶大小增加、占位效应明显;缺乏脊髓受累表现(MS有80%会受累,PCNSL<2%[6]);⑦脑脊液符合细胞学异常(克隆IgG基因重排),没有寡克隆带(PCNSL寡克隆区带阳性率27%,MS阳性率为98%[6])日本研究者TAKASONE等[17]认为早期脱髓鞘FDPPET显示部分病变表现出高代谢状态时也应高度怀疑PCNSL。
本例患者依据其前期的临床症状、影像学表现和治疗效果被初步诊断为MS,但患者短时间内病情反复并进行性加重,最后经术后病理确诊为PCNSL。在PCNSL确诊前,由该病引起的多灶性前哨脱髓鞘与特发性炎症脱髓鞘难以鉴别。
需要强调的是,“前哨病变”的诊断可以从临床的角度进行怀疑。当患者有增强造影剂的局灶性病变时,即使活检显示脱髓鞘改变,也应认真考虑PCNSL的诊断。这将决定治疗方案的改变,包括早期停用皮质激素,重复活检(在某些情况下),以及对患者进行密切的临床和影像学监测。患者如果在病变早期符合上述文献总结的PCNSL的典型特点[13],并结合病理学上广泛的T细胞、巨噬细胞浸润,轴突、髓鞘显著减少的表现,应考虑PCNSL的诊断。激素治疗可能会使PCNSL病灶变小或者消失,如果激素治疗后MRI表现符合淋巴瘤特征,为改善预后、降低死亡率,即使缺乏病理学证据,也可经验性给予抗PCNSL治疗[18]。
最后该病例的误诊让我们再次思考:诊断标准的应用给临床诊断带来循证医学依据的同时往往使我们缺少很多思考,加上明显改善的治疗效果很容易让我们放松警惕,停止对疾病寻根溯源的探索,所以造成早期辅助检查不够完善(未行脑脊液IgG寡克隆区带检查,无早期MRI强化检查),给疾病的进一步鉴别诊断带来困难;所以疾病诊断初期完善的辅助检查,治疗过程的明察秋毫,见微知著,治疗效果多方面的评价对减少疾病的漏诊和误诊至关重要。
猜你喜欢
中国实验诊断学(2023年5期)2023-05-26 01:32:42
中华耳科学杂志(2022年1期)2022-11-24 15:09:22
生物化学与生物物理进展(2022年11期)2022-03-02 11:34:48
医学新知(2019年4期)2020-01-02 11:04:02
党的生活(黑龙江)(2018年9期)2018-10-17 01:24:24
益寿宝典(2018年1期)2018-01-27 01:50:24
纺织科学研究(2017年6期)2017-07-03 12:14:26
大众健康(2017年1期)2017-04-13 09:01:04
当代医药论丛(2017年22期)2017-04-12 06:30:12
国防(2016年12期)2017-01-10 06:31:50
