
基于HPLC指纹图谱的当归不同干燥品质量研究△
2019-04-08 06:29:12李旭李成义陈杰焦彦斌强正泽李波王明伟
中国现代中药 2019年3期
李旭,李成义*,陈杰,焦彦斌,强正泽,李波,王明伟
1.甘肃中医药大学 药学院,甘肃 兰州 730000;2.甘肃天士力中天药业有限责任公司,甘肃 定西 748100
当归来源于伞形科植物当归Angelicasinensis(Oliv.)Diels的干燥根,始载于《神农本草经》。味甘、辛,性温。具有补血活血、调经止痛、润肠通便的功效[1]。当归所含化学成分复杂多样,主要涉及挥发油、有机酸、多糖和黄酮等成分[2-6]。现行的当归质量标准由辨别真伪优劣的定性指标(如采收期、产地、性状等)和定量指标(如水分、挥发油、阿魏酸含量等)组成,控制的是当归某部分组分,忽略了多组分的协同作用。中药指纹图谱基于对中药物质群整体作用的认识,具有信息量大、特征性强、整体性和模糊性等特点,与中医整体观相适应,成为中药的“化学条码”,是实现鉴别中药真伪优劣的可行模式[7-9]。
中药材产地加工是中药材生产和品质形成必不可少的重要环节,这一环节的实现使药材质量得以保证,且便于贮存、运输[10-11]。传统干燥加工方法简单易行,但往往干燥周期长、费时费力,无标准可循,导致加工中药材品质参差不齐。现代干燥加工方法的涌现,为中药材干燥加工增添了新的内容。本实验采用HPLC指纹图谱技术,建立了当归HPLC指纹图谱,通过相似度评价、主成分分析和聚类分析对当归不同干燥品进行了分析,以期为评价当归干燥方法提供依据。
1 仪器与材料
1.1 仪器
Agilent 1100高效液相色谱仪(美国Agilent公司),KQ-500DE型数控超声波清洗器(昆山市超声仪器有限公司),FW177型中草药粉碎机(天津市泰斯特仪器有限公司),CAV214C型电子天平(美国奥豪斯公司),BT25S型1/100 000电子天平(北京赛多利斯仪器有限公司),Milli-Q Century超纯水机(德国默克公司)。
1.2 材料
对照品阿魏酸、绿原酸(中国食品药品检定研究院,批号分别为110753-201718,110753-201718);洋川芎内酯A、正丁基苯酞、Z-藁本内酯(北京北纳创联生物技术研究院,批号分别为62006-39-7,6066-49-5,4431-01-0);乙腈为色谱纯;其余为分析纯。
当归新鲜样品采自当归主产区甘肃岷县,经甘肃中医药大学药学院李成义教授鉴定为伞形科植物当归Angelicasinensis(Oliv.)Diels的根。将采集的新鲜当归根做不同的干燥处理,样品信息见表1。

表1 当归样品信息表
2 方法与结果
2.1 色谱条件[12]
色谱柱为TC-C18(2)色谱柱(250 mm×4.6 mm,5 μm),流动相为乙腈(A)-1%乙酸水溶液(B),梯度洗脱(0~25 min,5%~25%A;25~40 min,25%~55% A;40~60 min,55%~80% A),流速0.6 mL·min-1,检测波长280 nm,柱温30 ℃,进样量10 μL。
2.2 对照品溶液的制备
取阿魏酸、绿原酸、洋川芎内酯A、正丁基苯酞对照品适量,精密称定,加甲醇溶解于10 mL容量瓶中,配制成质量浓度为0.21、0.25、0.14、0.06 mg·mL-1的阿魏酸、绿原酸、洋川芎内酯A、正丁基苯酞混合对照品溶液;精密称定Z-藁本内酯对照品15.23 mg,置于5 mL容量瓶中,加甲醇超声溶解,定容至5 mL,即为Z-藁本内酯对照品溶液。
2.3 供试品溶液的制备
取当归药材粉末约1.0 g,精密称定,置具塞锥形瓶中,精密加入20 mL 50%甲醇水,密塞,称定重量。超声提取40 min后冷却,再以50%甲醇水补足减失的重量,摇匀,静置。取上清液过0.45 μm微孔滤膜,即为供试品溶液。
2.4 方法学考察
2.4.1 精密度试验 取S11号样品约1.0 g,精密称定,按“2.3”项下方法制备供试品溶液,按“2.1”项下色谱条件测定,连续进样6次,检测指纹图谱。各共有峰的相对峰面积RSD均小于2.56%,相对保留时间RSD均小于0.3%。将连续进样6次得到的色谱图导入国家药典委员会“中药色谱指纹图谱相似度评价系统”(2012.1版)进行相似度分析,结果显示相似度均大于0.990,表明仪器精密度良好。
2.4.2 稳定性试验 取S11号样品约1.0 g,精密称定,按“2.3”项下方法制备1份供试品溶液,按“2.1”项下色谱条件测定,分别在制备后0、2、4、8、12、24 h进样检测,测得各共有峰的相对峰面积RSD均小于2.93%,各共有峰的相对保留时间RSD均小于0.25%。将样品图谱导入“中药色谱指纹图谱相似度评价系统”(2012.1版)进行相似度分析,结果显示相似度均大于0.990,表明供试品溶液在24 h内稳定性良好。
2.4.3 重复性试验 分别取S11号样品6份,每份约1.0 g,精密称定,按“2.3”项下方法制备供试品溶液6份,分别进样检测。测得各共有峰的相对峰面积RSD均小于2.67%,各共有峰的相对保留时间RSD均小于0.2%。将得到的图谱导入“中药色谱指纹图谱相似度评价系统”(2012.1版)进行相似度分析,结果显示相似度均大于0.990,表明方法重复性良好。
2.5 当归药材指纹图谱研究
2.5.1 指纹图谱的建立 取42批当归药材按“2.3”项下方法制备供试品溶液,按“2.1”项下色谱条件测定,得到当归不同干燥品的液相色谱图。取绿原酸、阿魏酸、洋川芎内酯A、正丁基苯酞、Z-藁本内酯对照品按“2.2”项下制备对照品溶液,按“2.1”项下色谱条件测定,得到绿原酸、阿魏酸、洋川芎内酯A、正丁基苯酞的混合对照品的液相色谱图和Z-藁本内酯对照品的液相色谱图,见图1。将42批当归药材的液相色谱图导入国家药典委员会“中药色谱指纹图谱相似度评价系统”(2012.1版),设S41为参照图谱,采用中位数、时间窗为0.1的方法,经多点校正后对色谱峰进行全谱图匹配,见图2,并生成共有模式的对照指纹图谱,见图3。
2.5.2 共有峰的标定 根据所建立的当归指纹图谱,在对照指纹图谱上共标定出24个共有峰,见图3。通过与对照品溶液色谱图的比对,指认了其中的4个共有峰,分别为7号峰绿原酸、11号峰阿魏酸、22号峰洋川芎内酯A、24号峰Z-藁本内酯。经比对,22号峰后紧接着出的峰(52.4 min)为正丁基苯酞,在某些样品中未检测到该峰。其中11号峰阿魏酸分离度高,是2015年版《中华人民共和国药典》规定的当归指标性成分且峰面积较大,故以其作为参照峰计算其余各峰的相对保留时间和相对峰面积。结果发现不同干燥处理的样品相对保留时间RSD均<3%,而相对峰面积的RSD偏大,说明当归不同干燥品的共有成分相对含量差异较明显。
2.5.3 相似度评价 采用“中药色谱指纹图谱相似度评价系统”(2012.1版)对 42 批样品的指纹图谱数据进行分析,计算 42批当归和对照指纹图谱之间的相似度,结果见表2,相似度均大于0.920,表明当归不同干燥品间相似度较好。
2.5.4 当归不同干燥品HPLC指纹图谱比较 根据计算得到的共有峰相对保留时间和相对峰面积分析,当归不同干燥品差异主要集中在峰面积上,即化学成分的种类差别不大,但各共有峰对应化学成分的含量均有明显差异。值得注意的是,S40~S42(阴干样品)中正丁基苯酞峰(出峰时间52.4 min)未检测到,可能是因为阴干过程耗时长,该成分损耗较大;S37~S39(熏干样品)在13.2 min处比其他样品多出了1个峰,且峰面积较大,可能为传统熏干当归的特征峰。
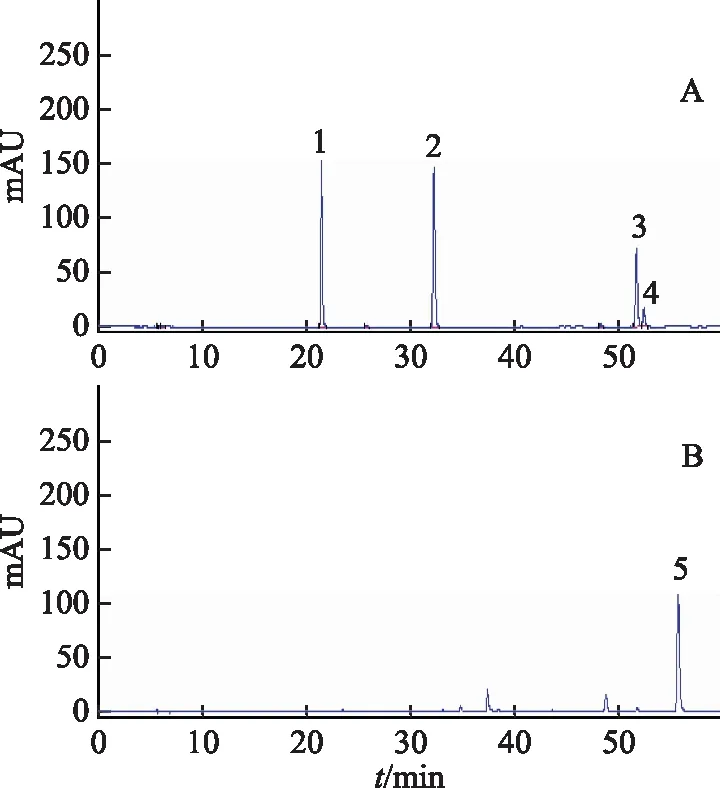
注:1.绿原酸;2.阿魏酸;3.洋川芎内酯A;4.正丁基苯酞;5.Z-藁本内酯。图1 混合对照品HPLC图(A)和Z-藁本内酯对照品HPLC图
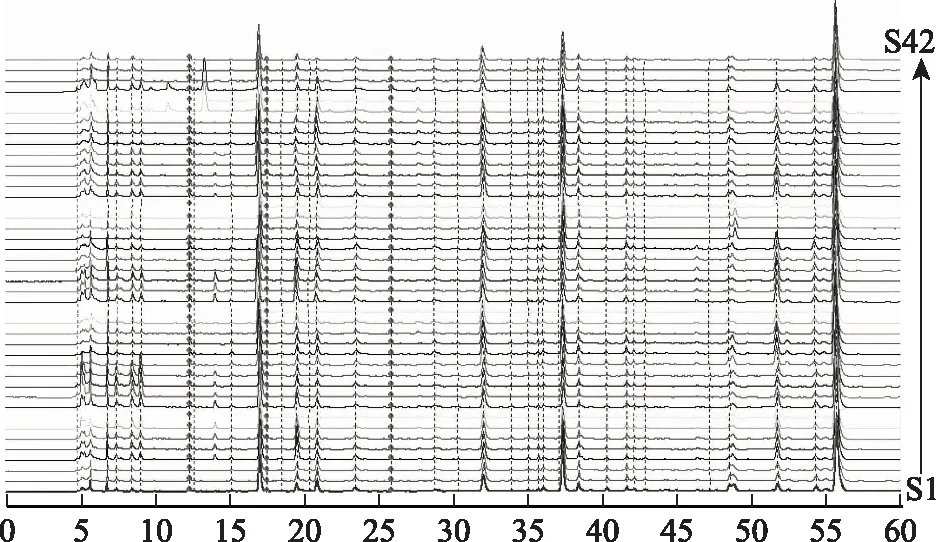
图2 42批当归药材叠加指纹图谱
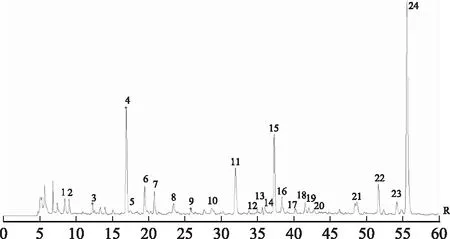
图3 对照指纹图谱
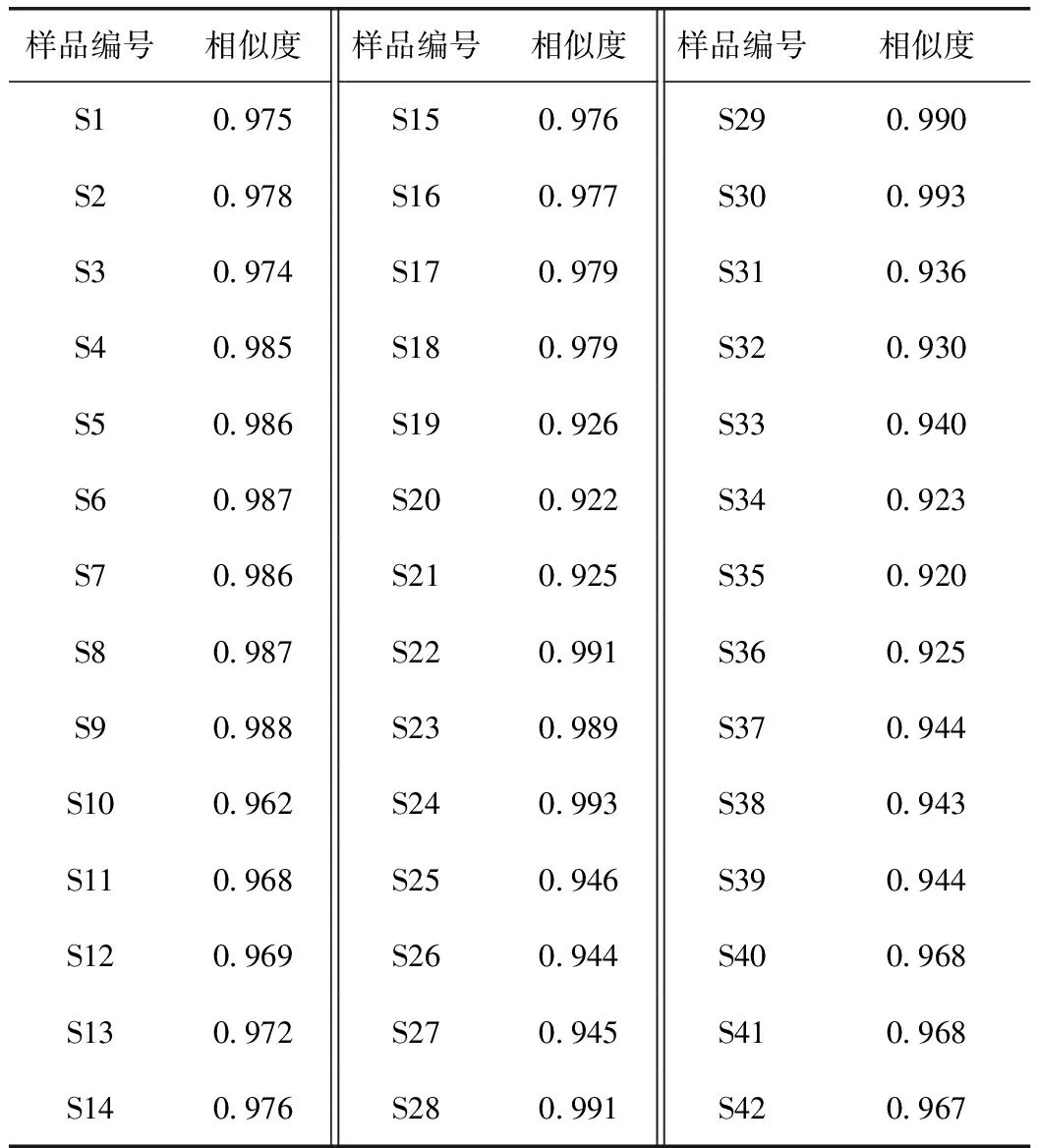
表2 不同干燥品指纹图谱相似度评价
2.6 主成分分析
将共有峰峰面积导入SPSS 21.0软件,对24个共有峰进行主成分分析,得到24个共有峰的初始特征值和方差贡献率(表3)。前5个成分的方差累积贡献率达86.093%,特征根大于1,可以代表当归指纹图谱中24个共有峰的大部分信息。由因子载荷矩阵得到主成分载荷矩阵U,以特征向量U值作为系数,得到5个主成分的表达式,将原始变量标准化处理后计算得到Y1、Y2、Y3、Y4、Y5的值。以各公因子的旋转之前的方差贡献率为权重系数,计算当归不同干燥品的综合得分,Y=0.418Y1+0.186Y2+0.128Y3+0.077Y4+0.052Y5。由表4可知,排名靠前的样品有60 ℃微波干燥、减压干燥、60 ℃热泵干燥品。
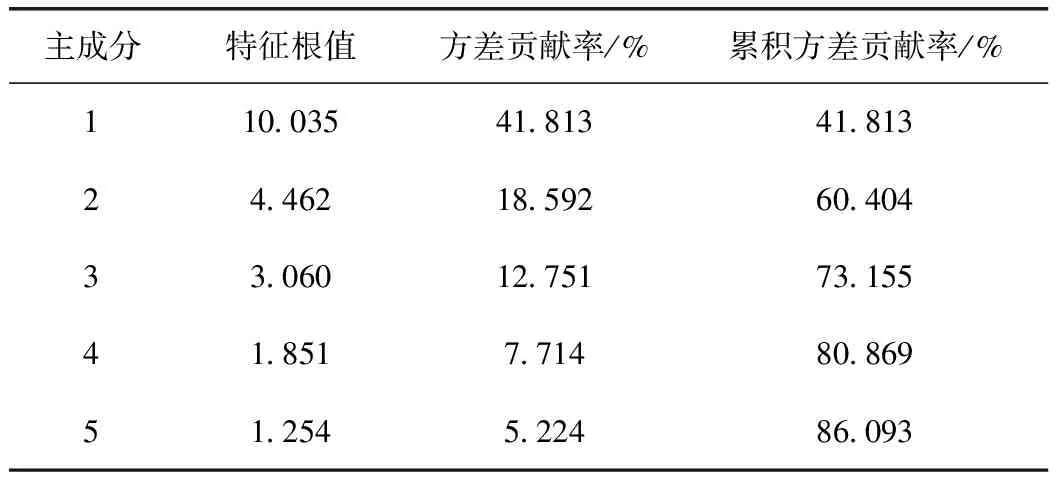
表3 主成分特征值及方差贡献率
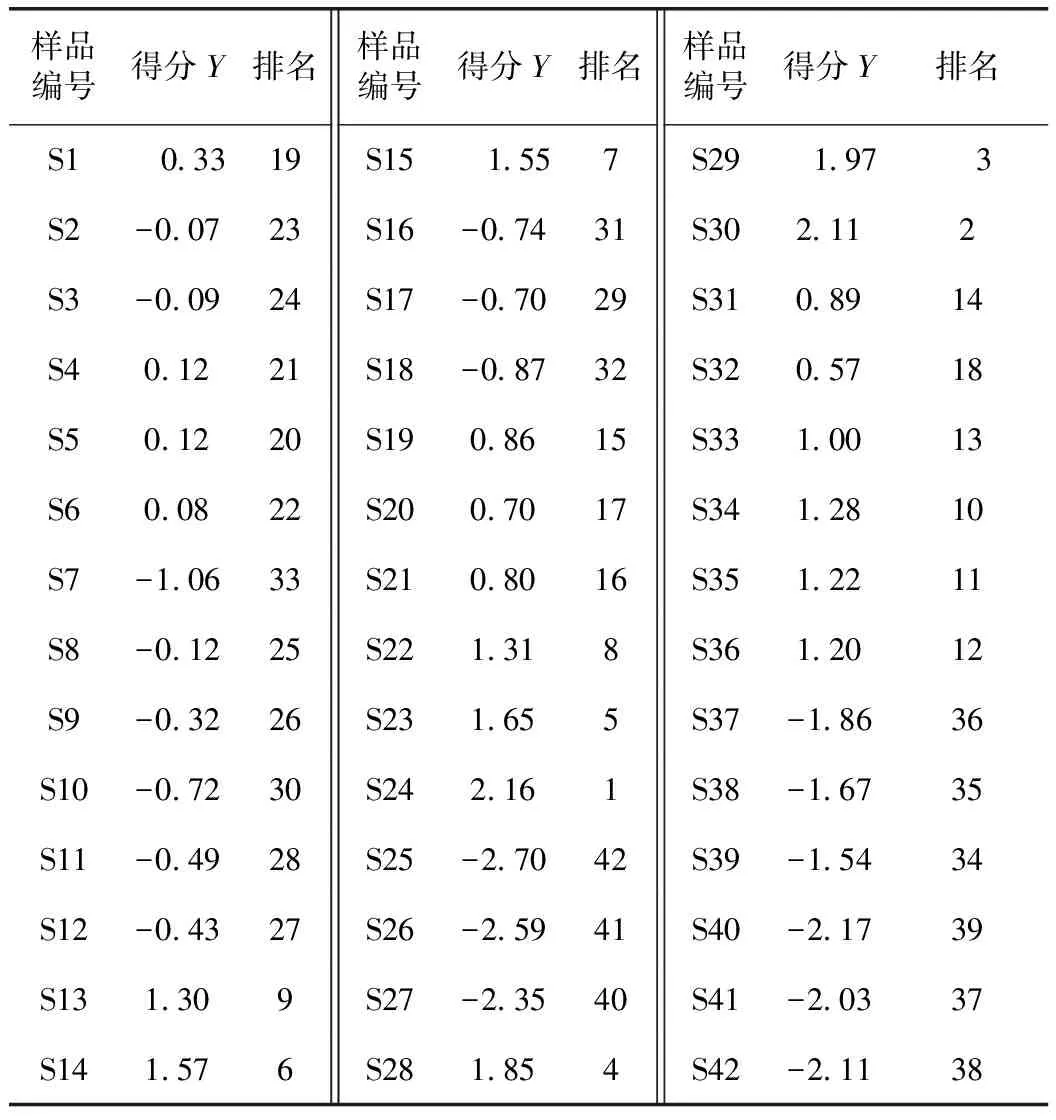
表4 当归不同干燥品综合得分及排名
2.7 聚类分析
以主成分得分作为变量,运用组间联接法进行系统聚类分析,结果见图4。当欧氏距离为25时,14批样品可分为两大类,S37、S38、S39聚为一类,其余样品聚为另一类,即熏干样品单独聚为一类,其余干燥方法得到的样品聚为一类。欧氏距离为18时,剩余干燥品聚为两类,阴干和晒干品聚为一类,其余聚为另一类。
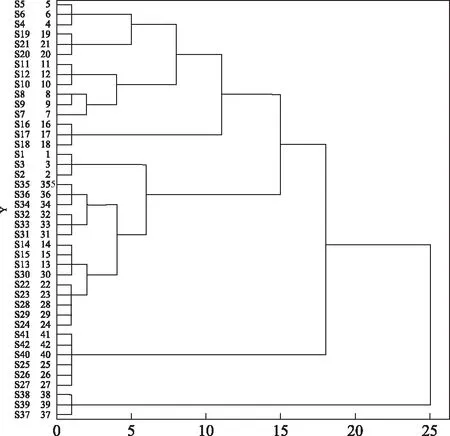
图4 当归指纹图谱聚类分析
3 讨论
为保证建立的当归不同干燥品HPLC指纹图谱出峰数目多、分离度好,本实验前期对色谱条件进行了考察。考察了甲醇-水、乙腈-水、乙腈-1%乙酸水三种流动相,发现以乙腈-1%乙酸水溶液为流动相分离效果最佳,出峰数目多,故选择乙腈-1%乙酸水溶液为流动相;考察了235、254、275、280、320 nm五个检测波长,发现在280 nm下,图谱基线平稳,色谱信息较为全面,故选择280 nm作为检测波长;考察了1.0 mL·min-1和0.6 mL·min-1两个流速对色谱图的影响,发现0.6 mL·min-1流速下分离效果更佳,故选择0.6 mL·min-1作为最佳流速。
本实验运用相似度评价、主成分分析和聚类分析方法对当归不同干燥品HPLC指纹图谱进行分析。相似度分析结果显示,各样品间相似度均大于0.920,表明当归不同干燥品间相似度良好。主成分分析结果显示60 ℃微波干燥、减压干燥、60 ℃热泵干燥样品排名靠前,而评分较低的的几批当归为晒干、阴干、熏干品。当归为油质类药材,减压干燥能最大程度的减少干燥过程中化学成分的损失,传统阴干、晒干、熏干品干燥时间长,干燥效率低,在长期干燥的过程中,伴随失水其化学成分的含量明显降低。本实验结果排名靠前的样品有60 ℃干燥品,跟相关文献报道[13-14]有出入,基于此研究结果,可能的原因是高温干燥下鲜药材快速失水,致使药材表面形成“硬皮”,药材内部失水变缓,化学成分损失亦减缓。
2015年版《中华人民共和国药典》描述的当归干燥方法为熏干,历史上鲜有本草记载以熏干为当归干燥方式。聚类分析发现,熏干样品单独聚为一类,其余干燥品聚为一类,说明传统阴干、晒干和现代干燥方法得到的当归药材质量较为一致,可为现代干燥技术在当归药材初加工的应用提供一定科学依据。
猜你喜欢
中国中医药现代远程教育(2024年8期)2024-03-27 09:13:06
西北植物学报(2022年12期)2022-02-13 10:06:48
云南化工(2021年5期)2021-12-21 07:41:12
天然产物研究与开发(2019年1期)2019-03-01 05:41:08
中成药(2018年11期)2018-11-24 02:56:46
中成药(2017年10期)2017-11-16 00:50:42
首都食品与医药(2017年21期)2017-11-03 06:45:32
应用化工(2014年3期)2014-08-16 13:23:50
应用技术学报(2014年1期)2014-02-28 14:52:13
首都食品与医药(2013年9期)2013-10-19 04:42:36
