
基于氧化还原共沉淀法制备的Mn-Ce催化剂及其低浓度甲烷燃烧催化性能
2019-04-03 08:15:54,,,,,,
燃料化学学报 2019年3期
, , , , , ,
(华中科技大学 煤燃烧国家重点实验室, 湖北 武汉 430074)
近年来,来自固定源和移动源的温室气体成为主要的环境问题,因而受到研究者的广泛关注[1]。由于甲烷大气温室效应是二氧化碳的21倍(100年计),且其对温室效应贡献率占温室气体总贡献率的18.6%,因而甲烷被认为是最严重的大气温室污染物之一[2,3]。煤矿瓦斯是全球大气甲烷的主要来源,约占全球甲烷排放总量的8%[4]。由于乏风瓦斯中甲烷浓度为0.1%-1.0%(甲烷化学性质稳定),而氧浓度数倍于甲烷浓度,其他乏风瓦斯处理技术如变压吸附、膜分离、低温分离等均存在爆炸,高成本等缺点。而催化燃烧因成本较低,而应用广泛,但需要寻找低温着火性能好,高温热稳定性强的催化剂[5, 6]。
尽管贵金属催化剂在甲烷低温催化燃烧和催化选择性方面有着良好的表现,但由于其全球储量较少[7]、成本偏高,同时其抗水抗硫性能较差[8]。因而,价格相对较低、含量丰富的过渡金属及其他非贵金属受到研究人员的广泛关注[9, 10]。
Zhang等[11]采用共沉淀法制备Mn-Ni催化剂用于低浓度甲烷催化燃烧,其将Mn含量为0.13时催化剂的高活性归结于Mn-Ni固溶体导致的Mn离子的广泛分布以及Ni的高的氧空位。Hu等[12]研究表明,Ce掺杂Ni催化剂不仅提高了催化剂的还原性,同时增加了晶格氧浓度,这有利于实现催化剂催化氧化能力。
以上研究结果表明,金属Mn、Ce具有较高的氧化还原能力,目前,Mn-Ce催化剂被广泛应用在NH3-SCR[13]、CWAO[14]及氨氧化[15]等低温催化领域,在甲烷催化领域应用较少[16-20]。这是因为Mn-Ce催化剂的氧化还原能力强烈依赖于催化剂颗粒尺寸、表面结构以及活性位点的分布情况。而催化剂的制备方法与组成成分对催化剂的表面结构和活性位点分布有着重大影响[9]。
目前,Mn-Ce催化剂的制备主要包含共沉淀法、溶胶凝胶法、燃烧法、活性剂辅助沉淀法及水热法[21],相较于其他制备方法,共沉淀法因为其工艺简单,对制作环境要求不高,因而在工业上有着广泛的应用,但沉淀剂的加入会导致局部pH值过高,破坏动力学沉淀平衡,从而产生团聚导致催化剂成分分布不匀。而经改进后的氧化还原共沉淀法则能够弥补沉淀剂导致的pH值局部波动的缺点,其通过控制滴加速率,达到控制沉淀的目的,进而使得催化剂减少团聚,组成更加均匀,同时氧化还原共沉淀法能产生更多的α-MnO2[22],而α-MnO2对甲烷的催化活性明显高于其他晶形的MnO2[23]。目前,关于氧化还原共沉淀法制备Mn-Ce催化剂用于甲烷催化燃烧方面的研究报道极少。
本研究采用氧化还原共沉淀法制备了一系列Mn-Ce催化剂,开展了低浓度CH4气体催化燃烧活性实验研究,进一步对催化剂物理化学性能进行表征。通过上述工作,对Mn-Ce催化剂的组成成分进行了优化研究,该参数对甲烷活化过程十分重要。通过对该参数的优化,研究催化剂活性变化的内在机理,从而有助于理解该催化剂对甲烷的氧化过程。
1 实验部分
1.1 催化剂的制备
本研究通过氧化还原法制备一系列不同比例的Mn-Ce催化剂,该制备方法具体见文献[24, 25]。即将一定量的硝酸铈铵((NH4)2Ce(NO3)6AR 99.0%)和高锰酸钾(KMnO4GR)混合溶液以一定速率加入到被磁力搅拌器以一定速率搅拌的硝酸锰(Mn(NO3)2AR 50% in H2O)溶液中,同时滴加一定浓度的氢氧化钾(KOH ACS)溶液,从而使得混合溶液pH值为4.5±0.3。将沉淀用抽滤瓶过滤,用超纯水和无水乙醇清洗沉淀三次,然后在100 ℃鼓风干燥机中干燥12 h,将干燥后的样品放置在500 ℃的马弗炉中焙烧6 h,待样品冷却后用研钵研磨并筛分出40-60 目固体颗粒,即所制得的催化剂。根据催化剂中Mn的含量(如30%、50%、70%、90%),将所制得的催化剂在下文中将表示为Mn30、Mn50、Mn70、Mn90。作为对比,将高锰酸钾(KMnO4GR)溶液加入硝酸锰(Mn(NO3)2AR 50%(质量分数) in H2O)溶液中,并加入KOH溶液保持pH值为4.5±0.3,经过上述相同的处理过程,制得MnOx催化剂表示为Mn100。
1.2 催化剂的表征
使用ASAP2000型氮吸附仪(美国Micromeritics公司)测定样品的比表面积(BET)及孔分布(BJH)参数。每次实验将约200 mg样品在300 ℃,6.7 Pa条件下脱气2 h,测试采用液氮(77 K)等温吸附法,通过Brunner-Emmet-Teller(BET)和Barret-Joyner-Halenda(BJH)方程计算得到比表面积和平均孔径。
使用X’pert3Powder型X射线衍射仪(XRD)(荷兰PANalytical公司)完成表面物相分析,分析扫描为5°-85°,每步0.02°。
使用EAGLE III型 X 射线荧光探针(XRF)( 美国 EDAX公司) 进行表面元素分析,检测催化剂表面的金属元素的含量,分析使用参数为30 kV、500 μA,检测电极为Rh电极。
使用AXIS-ULTRA DLD-600W型 X射线光电子能谱仪(XPS)(日本岛津-Kratos公司)分析催化剂表面元素价态,及元素结合能。使用XPSPEAK4.1软件对样品的能谱图进行Gauss拟合,对样品表面的元素进行定性分析及定量计算。
1.3 催化剂的活性评价
催化剂活性评价在自搭的微型固定连续流动反应床(id= 6 mm)上完成,实验装置示意图见图1。管内装有200 mg催化剂(40-60目),反应气体组成成分(体积分数)为1% CH4、10% O2和N2平衡,反应气流总量为100 mL/min,质量空速比(WHSV)为30000 mL/(h·g)。在实验前,首先对催化剂进行活化预处理(在400 ℃下通入100 mL/min空气吹扫30 min,然后切换至100 mL/min的N2吹扫30 min),冷却至室温后,开始活性检测,将固定床出来的反应尾气通过GC9700 II(FID,TCD检测器)检测剩余CH4、CO及CO2浓度,结果发现出口CO可以忽略不计,反应的CH4完全转化为CO2。实验测试时,当加热炉温度稳定到测定温度30 min后,开始至少三次采样(取RSD<0.3的三次结果平均值)。甲烷转化效率定义为:
(1)
式中,Cin和Cout分别代表反应床入口和出口的甲烷浓度(g/mL)。
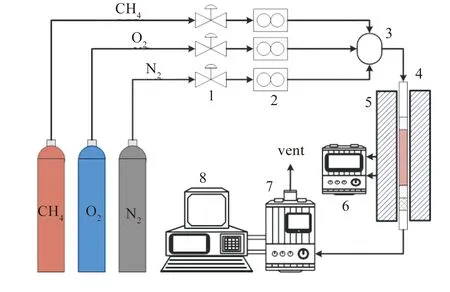
图 1 催化剂活性实验装置示意图
2 结果与讨论
2.1 催化剂的活性
图2为不同Mnx(x=30、50、70、90、100)催化剂的甲烷转化率结果。

图 2 不同催化剂的甲烷转化率
由图2可知,本实验所有催化剂在560 ℃时甲烷转化率都在90%以上,表明催化剂在高温区域具有较好的催化活性。本研究以文献中常用的甲烷转化率50%时的温度t50来评价催化剂的活性,由图2可知,Mn90催化活性最佳(t50=~446 ℃)。Mn含量从30%增加到90%过程中,随着Mn含量的增加催化剂的活性逐渐上升。随后,继续增加Mn含量到100%,催化剂活性明显下降(t50=~456 ℃)。
Mn作为一种过渡元素,全球储量明显高于稀土元素Ce[7]。对比文献中的使用Mn-Ce催化剂用于低浓度甲烷气流的催化燃烧结果发现,本研究结果在Mn/Ce比和催化剂活性两方面存在优势。Fiuk等[16]使用共沉淀法制备MnCe催化剂,其催化剂的最佳Mn/Ce比为7∶3,低于本实验的最佳Mn/Ce比;Jing等[26]使用共沉淀法制备的MnCe催化剂的最佳Mn/Ce比为6∶4,也低于本实验的Mn/Ce比,同时该研究中催化剂的最佳活性(t50=~513 ℃)低于本实验的催化剂最佳活性;李树娜等[19]使用水热法制备的MnCe催化剂,尽管其最佳活性(t50=~440 ℃)与本实验相当;但其催化剂Mn/Ce比为1∶9,低于本研究的Mn/Ce比;Zhang等[18]使用溶胶凝胶法制备的MnCe催化剂最佳Mn/Ce比为5∶5,也低于本实验的Mn/Ce比。
Mn90和Mn100相比,Ce的掺入明显地提高催化剂的活性,这表明,MnCe之间发生强烈的交互作用,促进催化剂的活性提高。本研究将从催化剂物理化学性质方面研究探索催化剂Mn/Ce比与文献结果差别较大的原因。
2.2 BET分析
表1为五种催化剂MnOx(x=30、50、70、90和100)的比表面积、孔容与平均孔径。由表1可知,随着Mn含量的逐渐增加,催化剂的比表面积呈现先上升后下降的趋势,这表明存在最佳Mn含量使得催化剂的比表面积等参数最大,Ce的掺杂使得催化剂比表面积和孔容明显提高,这表明金属掺杂有利于催化剂比表面积的增大,这与文献[24]结论一致。催化剂Mn50、Mn70和Mn90三种催化剂的比表面积、孔容及平均孔径相差不大,且明显大于其他两种催化剂。较大的催化剂比表面积、孔容及孔径有利于催化剂活性位点的暴露,这都有利于催化剂活性的提升。

表 1 不同催化剂的氮吸附表征结果
Mn100的比表面积、孔容等明显小于Mn30、Mn50等双金属催化剂,这表明一定量的Ce掺杂有利于提高催化剂物理属性比表面积、孔容等物理性质[27]。
2.3 XRD分析
图3为五种催化剂MnOx(x=30、50、70、90和100)的 XRD 谱图。
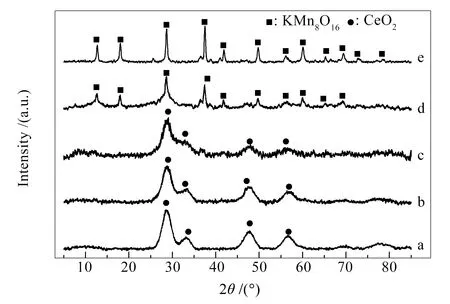
图 3 不同催化剂的 XRD 谱图
由图3可知,Mn/Ce比较低时,CeO2晶体(JCPDS 34-0394)呈现了28.56°、33.08°、47.48°、56.33°、69.40°和79.07°的特征衍射角形成的特征峰。随着Mn/Ce比逐渐增加到7∶3,所有峰的强度逐渐降低,半峰宽逐渐变宽,各个峰发生融合,这表明,Mn、Ce相互作用得到增强[16],同时XRD谱图上仅呈现CeO2(JCPDS 34-0394)的峰,没有显示Mn的峰,这表明,MnOx以非晶形式存在。随着Mn/Ce比继续增大到9∶1时,KMn8O16晶体(JCPDS 29-1020)开始呈现12.75°、18.06°、28.74°、37.62°、42.03°、49.89°和60.24°的特征衍射角形成的特征峰,而CeO2的特征峰基本消失。由于物相KMn8O16在其他Mn-Ce甲烷催化剂中尚未检测到[16, 18-20],在氧化还原法制备Mn-Ce催化剂用于苯甲醇催化反应中出现[24],结合BET结果作者认为,Mn-Ce催化剂中物相KMn8O16有利于催化剂的活性提升。当Mn/Ce比为9∶1时,仅检测到少量CeO2的特征峰,表明CeO2高度分散在KMn8O16体系中,随着Mn含量进一步增加到100%,KMn8O16特征峰强度明显增加,这表明,在Mn90中部分Mn以非晶形式存在或者与Ce发生相互作用,结合图2结果,作者认为Ce的加入在一定程度上能够活化MnOx中不同Mn之间的电子传递,提高催化剂的催化活性[28]。
2.4 XRF分析
表2为五种催化剂MnOx(x=30、50、70、90和100)的XRF检测结果。XRF分析作为一种体相分析方法,检测出进入Mn、Ce氧化物晶格中的K(物理洗涤方法仅能去除催化剂表面的部分K)。

表 2 MnCe催化剂的XRF表征
由表2可知,随着锰含量的逐步上升,催化剂中的K摩尔含量呈现先下降后上升的趋势。当Mn/Ce比较小时,部分K进入过量的Ce晶格,导致催化剂K含量较高;随着Mn/Ce比增加,Mn、Ce相互作用增强,生成固溶体,导致K含量降低;随着Mn/Ce比进一步增加,物相KMn8O16生成,大量K进入该物相内部,导致K含量又重新增加。
XRF分析结果表明,催化剂表面的各原子物质的量比(Mn/Ceexp)与理论原料比(Mn/Ceth)并不相等,这与文献[29]研究一致。催化剂中各个元素之间存在相互作用,导致催化剂表面的Mn元素含量相对较少。文献研究表明,Mn2+(0.083 nm)、Mn3+(0.066 nm)和Mn4+(0.056 nm)离子半径均小于Ce3+( 0.103 nm) 和Ce4+(0.099 nm)的离子半径,部分Mn离子可能进入到了CeO2的晶格,从而形成Mn-Ce固溶体结构[27]。
2.5 XPS分析
为了确定催化剂表面元素价态,进一步分析最佳Mn/Ce比活性最高的原因,对实验催化剂进行了XPS检测。表3为催化剂表面O、Mn、Ce的统计结果。
图4为五个样品MnOx(x=30、50、70、90和100)的Mn 2p3/2轨道的XPS谱图。已有文献[23]及XRD分析结果表明,本研究所制催化剂中的Mn是以Mn4+、Mn3+共存的混合价态。一般认为Mn4+处于642.5-643.0 eV,Mn3+处于641.5-642.0 eV[22]。由图4及表3结果可以看出,随着Mn含量的逐渐增加,催化剂表面Mn4+的含量逐渐增加,这与XRD结果一致。Mn100中Mn4+/Mn3+的比值为7.67,与KMn8O16中Mn4+/Mn3+的比值相近,这也从侧面证明KMn8O16物相存在。文献[18]表明,Mn-Ce催化剂甲烷催化燃烧中Mn4+/Mn3+比值与甲烷燃烧速率成正相关关系。催化剂表面高浓度的Mn4+离子能提高催化剂氧化CH4的能力,有助于提高催化剂对低浓度甲烷的催化反应活性。

表 3 Mn-Ce催化剂表面的的元素组成

图 4 不同催化剂的Mn 2p3/2 XPS谱图
图5为四个样品MnOx(x=30、50、70、90)的Ce 3d的XPS谱图,其中,u‴、u″、u和v‴、v″、v六个峰面积之和对应Ce4+离子含量,而u′和v′两个峰面积之和对应Ce3+离子含量。

图 5 不同催化剂的Ce 3d XPS谱图
由图5可知,催化剂中Ce3+含量较少,催化剂中的Ce主要以4+形式存在。由表3可知,随着Mn含量的增加,催化剂中Ce3+的比例从0.11增加到0.28,这和催化剂活性趋势一致。Ce3+浓度的增加有利于产生氧空位及形成不饱和化学键,这对提高氧的吸附及迁移有促进作用,进而有利于提高催化剂的氧化还原能力[30]。
图6为五个样品MnOx(x=30、50、70、90和100)的K 2p的XPS谱图。由图6可知,样品均在292.2-293.3 eV和295.0-295.9 eV处存在两个峰。从图6可以看出,随着Mn含量的逐渐增加,K的键能向低能量方向移动,这表明催化剂表面的K的键能与Mn/Ce比存在强烈的相关性。文献[31]研究表明,KMn8O16中的K电子云与Mn电子云相互重叠,降低了Mn-O-Mn之间的键能,有利于催化过渡态能垒的降低,从而促进催化剂活性提高。

图 6 不同催化剂的K 2p XPS谱图
图7为五个样品MnOx(x=30、50、70、90和100)的O 1s的XPS谱图,在527-535 eV出现明显的双峰结构。

图 7 不同催化剂的O 1s XPS谱图
在529.41-529.83 eV处出现的峰归于晶格氧(用Oα表示),在531.4-531.5 eV处出现的峰归于吸附氧(用Oβ表示),在533.0-533.1 eV处出现的峰归于吸附水中的羟基氧(用Oω表示)[22]。文献[31,32]研究表明,Mn基催化剂在有机物催化燃烧中,遵循MvK(Mars and van Krevelen)机理,因此,更多的晶格氧有利于提高催化剂的活性。由表3还可知,随着Mn含量的逐渐增加,晶格氧在活性氧中的占比Oα/(Oα+ Oβ)呈现先上升后下降的趋势。其中,Mn90的晶格氧含量最高,这与催化剂活性实验结果相一致。
XPS谱图结果表明,较高的Mn含量能够提高催化剂中Mn4+的比例,增加Ce3+的含量以及提高晶格氧的浓度,同时,高的Mn含量能够增强Mn-K之间的相互作用,一定量的Ce能提高催化剂中晶格氧的含量,以上这些都有利于催化剂活性的提升。
3 结 论
本研究采用氧化还原共沉淀法,开展了系列Mn-Ce催化剂的(Mnx,x=30、50、70、90和100)制备与物理化学性能表征,以及低浓度CH4气体催化燃烧的实验研究。结果表明,Mn90催化剂在质量空速比为30000 mL/(h·g)工况下,催化活性最佳(t50=~446 ℃);恰当的Mn/Ce比能够增加催化剂比表面积、孔容、孔径等参数,产生KMn8O16物相,提高催化剂中Mn4+含量,氧空位及晶格氧浓度,从而使催化剂具有更佳的催化剂活性;将Mn90和Mn100对比可以看出,适量的Ce掺杂能够提高催化剂的晶格氧浓度,提高催化剂的比表面积和孔容等物理性质。
猜你喜欢
能源化工(2022年3期)2023-01-15 02:26:43
军民两用技术与产品(2021年10期)2021-03-16 06:05:08
水上消防(2020年1期)2020-07-24 09:26:02
净水技术(2020年12期)2020-02-16 11:26:12
数学物理学报(2019年5期)2019-11-29 07:46:50
疯狂英语·新读写(2018年3期)2018-11-29 22:37:11
北京航空航天大学学报(2017年9期)2017-12-18 07:12:18
数学物理学报(2017年5期)2017-11-23 07:51:09
宿州学院学报(2016年7期)2016-08-08 09:51:53
潍坊学院学报(2016年6期)2016-04-18 13:56:55
