
对抗生素注射剂一致性评价/再评价的思考
2019-03-29 05:21:04胡昌勤
中国抗生素杂志 2019年3期
胡昌勤
(中国食品药品检定研究院,北京 102629)
2015年8月18日,国务院印发的《关于改革药品医疗器械审评审批制度的意见》,将“提高仿制药质量,加快仿制药质量一致性评价”作为改革药品审评审批制度的5大目标之一;以2016年3月5日国务院办公厅发布的《关于开展仿制药质量和疗效一致性评价的意见》为节点,国内以生物等效为目标的口服制剂一致性评价工作蓬勃开展; 2017年12月CED一致性评价办公室又发布了《已上市化学仿制药(注射剂)一致性评价技术要求(征求意见稿)》,揭开了注射剂仿制药一致性评价的大幕。抗生素药物在注射剂中所占的比例较大,我们曾对国内市场抗生素药品的质量现状进行过分析,探讨国产仿制药与原研药品的主要质量差距[1],本文针对抗生素注射剂的特点,进一步探讨抗生素注射剂一致性评价中的关键点及评价策略。
1 注射剂一致性评价的主要关注点
据文献报道[2],日本有17.2%的医生和37.2%的患者不愿意使用仿制药,其原因主要与疗效和安全性的差异有关,如仿制的万古霉素和替考拉宁注射剂,其在实际治疗中抗菌活性较原研产品分别低于14.6%和17.3%。因此,保证仿制药与原研制剂在治疗中具有相同的有效性和安全性是仿制药一致性评价应重点解决的问题。
对口服制剂,目前人们普遍将生物等效性作为评价仿制药与参比制剂是否具有临床一致性的关键指标。但对于注射剂,特别是水溶性注射剂,其生物等效性不是影响临床等效的主要原因,有哪些关键因素可能影响国产抗生素注射剂的临床一致性呢?
抗生素药物按其结构通常分为β-内酰胺类(包括青霉素与头孢菌素)、氨基糖苷类、大环内酯类、喹诺酮类、糖肽类等,评价抗生素类药物临床有效性的最理想参数是PK/PD(pharmacokinetic/pharmacodynamic)。由于不同类抗生素的抗菌作用机理不同,用于评价临床有效性的关键PK/PD参数也不相同。对β-内酰胺类抗生素,药物在体液中有效血药浓度(Ceff)的持续时间(T>MIC)是最关键参数;对氨基糖苷类抗生素和喹诺酮类抗生素,24h体内的AUC/MIC比值是最关键参数;而对大环内脂类和糖肽类等非浓度依赖性抗生素,如阿奇霉素和万古霉素等,AUC/MIC比值是最关键参数[3]。
Schito等[4]利用数学模拟的方法对多家注射用头孢曲松钠仿制药与原研制剂罗氏芬®(Rocephin®)在胸腔积液的有效游离药物浓度(Ceff)进行了预测,认为只有罗氏芬的Ceff可达到要求的浓度范围。国内临床反映,国产注射用头孢曲松钠较罗氏芬®起效约慢30~40min;由于头孢曲松钠为高血清蛋白结合药物,未成盐的头孢曲松酸较头孢曲松钠具有更高的蛋白结合率,而早期国产的头孢曲松钠成盐率相对较低,因此,与血清蛋白具有较高的蛋白结合率,导致早期国产仿制品在给药初期游离的药物浓度达不到Ceff,进而使得其起效时间延长[5]。
注射用美罗培南为美罗培南与碳酸钠的混粉。Agudelo等[6]利用豚鼠感染模型等证明,虽然仿制和原研注射用美罗培南在动物体内为生物等效,但部分仿制品的抗感染效果不如原研制剂;其原因是由于体内的肾毒性肽酶(DHP-I)对部分仿制药较原研制剂具有更强的水解能力。提示在临床应用中仿制药和原研注射用美罗培南可能出现不等效现象。由于国内注射用美罗培南仿制药和原研药的主要差异为产品中碳酸钠含量的不同,表现为仿制药溶液的pH明显高于原研制剂[7],这种差异是否会影响体内DHP-I酶的活性值得探讨。
利用GastroPlus软件,以万古霉素B为对象建立药代动力学模型,我们曾模拟多次给药条件下注射用盐酸万古霉素的人体内血药浓度曲线。结果显示,规格为500mg的注射用盐酸万古霉素,静脉滴注(给药时间为1h,间隔6h给药),健康成年男性体内的万古霉素血药谷浓度均达不到《中国万古霉素治疗药物监测指南》中推荐的10mg/L谷浓度;提示规格为500mg制剂药物剂量偏低。比较仿制药与原研制剂的装量,发现规格为500mg的原研制剂实际是按标示量的110%~115%投料,通常较国产制剂的装量约高13%[8];此外,由于万古霉素来源于生物发酵,产品中的小组分/杂质可以与万古霉素B竞争结合万古霉素靶位,导致疗效降低[9];而目前国产注射用盐酸万古霉素中万古霉素B的含量约较原研产品约低2%~3%[8];综上,注射用万古霉素由于装量及万古霉素B纯度的差异可导致国产仿制药较原研制剂的活性低约15%。
而对于多组分抗生素如庆大霉素、替考拉宁等,不同的组分对特定致病菌可能表现出不同的抗菌活性[10]。由于多组分抗生素的组分差异是客观存在的,当仿制药与参比制剂的组分比例不相同时,可能导致在特定的动物抗感染模型中表现出不同的抗感染效果[11],虽然其提示在对特定的病人治疗时候,可能出现疗效不一致现象;但这种差异可能在治疗其它致病菌的感染时呈现出完全不同的治疗效果。因此从整体角度,对多组分抗生素,仅依据个别致病菌建立的动物抗感染模型,比较仿制药与参比制剂的疗效一致性是不够严谨的。
对注射剂临床一致性评价的另一关注点是药物的安全性。原研注射用头孢孟多酯钠产品为头孢孟多酯钠与5%碳酸钠的混粉,而国内的仿制品中大多不含碳酸钠。注射用头孢孟多酯钠中加入适量的碳酸钠可以明显改善贮存过程中样品溶液的澄清度易变浑浊这一现象[12];而样品溶液的澄清度与药物的不良反应例数呈正相关,即不含碳酸钠的注射用头孢孟多酯钠较含5%碳酸钠的制剂的总不良反应例数更高,即含5%碳酸钠的注射用头孢孟多酯钠的安全性更高[13]。
综上,从临床应用的有效性与安全性出发,评价仿制药与参比制剂(原研制剂)工艺、处方差异的影响应是注射剂一致性评价的关键。
2 注射剂一致性评价与新仿制药研发策略的异同
仿制药一致性评价是对已上市的药品与参比制剂的临床疗效差异进行评价,通过处方/工艺等的改进等,使得产品的疗效与参比制剂一致。和药物研发中的新药仿制相比,虽然在研发/评价过程中都要从安全、有效、质量3方面与参比制剂进行比较, 但由于仿制药一致性评价是对“现实药品”的评估,而药品仿制是对“未来药品”的期望,因此,实现相同目标所采用的策略应不完全相同(图1)。不同于新仿制药通常需借助于“反向过程”的结果进行处方/工艺设计,国产仿制药的疗效与质量评价通常已经有大量的数据可以查阅,特别是历年的国家评价性抽验数据,其中蕴藏着大量的质量信息;一些被认为与原研药品处方、工艺有明显差异的品种,基本已进行过对比研究。因此,在进行仿制药一致性评价时,应首先通过对文献的调研,发现国产仿制药与参比制剂的关键质量差异,进而针对性的进行改进,而不应将精力主要放在与参比制剂的质量对比上。
案例1:注射用头孢曲松钠:头孢曲松钠(ceftriaxone sodiμm)为瑞士Roche公司在20世纪80年代研制的第三代头孢菌素,为结晶性粉末。其通过无菌分装工艺生产注射用头孢曲松钠,即制剂的质量主要取决于原料的质量。1991年头孢曲松钠及制剂在国内仿制成功。国内临床曾反映国产注射用头孢曲松钠较原研药品起效约慢30~40min。 2018年、2019年和2011年,中国食品药品检定研究院(以下简称中检院)对该品种曾组织过3次国家评价性抽验,发现国产头孢曲松钠的突出质量问题为溶液的澄清度差。据文献报道,国产头孢曲松钠的溶液澄清度[14-15],有关物质[15],成盐率[5]等方面与原研罗氏芬均存在差异, 成盐率偏低可导致头孢曲松钠具有更强的血清白蛋白结合率,可能导致给药初期血浆中的游离药物浓度偏低,影响药品的起效速率[5]。
头孢曲松钠分子含两个钠离子,3.5个水分子(图2a),湿品中头孢曲松含量的理论值为83.5%,水分子的理论值为9.52%;按无水物计,头孢曲松钠中头孢曲松含量的理论值为95.7%。头孢曲松钠结晶工艺的差异可导致产品成盐率偏低[16],进而使其与血浆蛋白的结合率增加,给药初期的游离药物浓度偏低,导致起效慢。采用粉末X射线衍射技术结合聚类分析手段,薛晶等[17]将头孢曲松钠的晶体特征分为不同的亚型,原研罗氏芬样品属第Ⅱ亚型;通过比较不同亚型样品的成盐率、水分等与结晶工艺相关的指标, 并比较加速实验过程中不同亚型样品晶格稳定性的差异与药用丁基胶塞相容性的差异,证明提高国产头孢曲松钠产品质量的关键应从控制产品的结晶工艺入手,保证终产品为第Ⅱ亚型的样品。

图1 仿制药一致性评价与新药仿制策略的比较Fig.1 Strategy comparison between quality consistency evaluation of marketing drug and new generic drug R&D
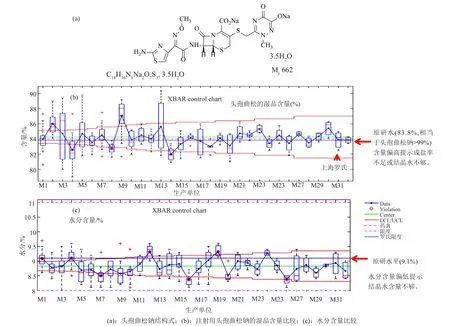
图2 国内不同企业注射用头孢曲松钠的质量对比Fig.2 Quality comparison of ceftriaxone sodiμm for injection in different domestic manufacture
对2009年的评价性抽验数据进行分析(图2b、图2c)。虽然全部产品均符合中国药典的规定,但不同企业产品的头孢曲松湿品含量和水分含量差异明显,且部分企业产品的批间差异较大,提示企业对产品的工艺控制能力较差;进一步分析发现,国产制剂的水分含量明显低于理论值(9.52%),且多数低于罗氏的产品(9.1%),提示部分产品中结晶水不足;湿品中头孢曲松的含量偏高,部分高于理论值(83.5%),提示部分产品存在成盐不完全现象;而上述差异又均与头孢曲松钠的结晶过程有关(未发表资料)。即提示头孢曲松钠的晶体特征是关键质量属性,对原料结晶工艺的控制是关键质控点。
综上所述,在对注射用头孢曲松钠进行一致性评价时,应以原料的结晶工艺及工艺控制为切入点,首先证明结晶工艺可以保证产品的晶体特征属于第Ⅱ亚型,再以产品湿品的头孢曲松含量和水分含量为指标对工艺过程进行优化,保证生产过程的一致性,降低批间差异。
3 注射剂一致性评价/再评价之一般策略
质量源于设计(QbD)理念在药品研发、生产、监管等领域已经被普遍接受。通常目标产品的质量可通过产品的关键质量属性(CQAs)进行表征[18-19]。因此,在进行仿制药一致性评价中,应首先对参比制剂的CQAs有深入的理解,在此基础上确定参比制剂生产过程中的关键原辅料属性(CMAs)和关键工艺参数(CPPs),建立CMAs 、CPPs和产品CQAs的关系。这不仅有助于揭示仿制药与参比制剂的主要质量差异,且可以针对性的对仿制药的处方、工艺进行评价;并通过对仿制药CMAs和CPPs的控制,实现对产品CQAs的控制,保证仿制药与参比制剂的一致性。只有当确认仿制药与参比制剂的CQAs基本一致时,对二者的药学指标进行系统的比较才有意义(图3)。
发现药品关键质量属性并与生产工艺相关联通常可以采用以下方法。
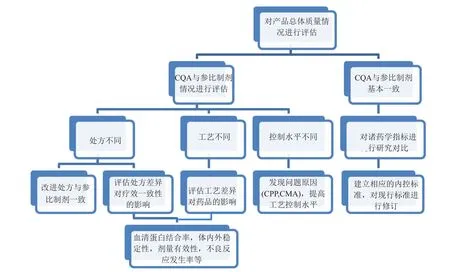
图3 进行仿制药一致性评价的一般策略Fig.3 General strategy for quality consistency evaluation of generic drug
3.1 利于国家评价性抽验数据结合文献进行分析
2015年,中检院通过对从2008年起历年国家评价性抽验涉及的15个注射用头孢菌素质量的系统回顾性分析[20],认为市场上注射用头孢菌素的突出质量问题是“溶液的澄清度与颜色”。通过对2008—2013年国家药品不良反应监测数据库的检索,将注射用头孢菌素的主要不良反应(ADR)报告与市场抽验结果相关联,发现注射用头孢曲松钠和注射用头孢唑林钠的严重不良反应比率在2011年后呈下降趋势,与二者在2010年后溶液的澄清度问题明显好转有直接相关。将“溶液的澄清度”作为CQAs,发现头孢菌素原料与胶塞的相容性直接影响注射剂的澄清度;使用镀膜/覆膜胶塞或选用质量较好的头孢菌素原料可以有效地阻断胶塞挥发物与头孢菌素的相互作用,改善头孢菌素与胶塞的相容性;而头孢菌素原料的结晶工艺是影响原料质量的关键工艺,优化CPPs是获得理想头孢菌素原料的关键,这也是目前国产仿制药与原研药品之间的最大差异。
对头孢菌素晶型、工艺与稳定性关系的研究揭示,头孢菌素可以形成不同晶型的晶体,也可以与水分子或溶剂相互作用形成不同的水合物或溶剂化物,通常具有不同空间结构的晶体(晶型/水合物)的固体稳定性不同[21-25]。如头孢哌酮钠具有A、B两种晶型,A晶型为三斜晶系,B晶型为单斜晶系;A晶型较B晶型更稳定。根据粉末X-衍射图谱特征衍射峰(峰位置、峰强度)的差异,A晶型样品又可分为多个亚晶型,亚晶型Ⅰ和亚晶型II在加速试验中表现出更高的稳定性,原研药品“先锋必”和“舒普深”中的头孢哌酮钠即为此亚晶型[23]。头孢米诺钠晶体含有7个水分子,根据单晶X-衍射结果,理想状态下每一晶格中含3个游离态的水分子和4个通过氢键与头孢米诺结合的水分子;但水分子在晶格中可以存在于多个位置,导致不同的结晶条件下可形成不同的亚晶型,在粉末X-衍射图谱中表现出细微的差异;不同亚晶型的头孢米诺钠样品具有不同的化学稳定性[25]。同样,头孢他啶含有5分子结晶水,不同工艺的头孢他啶产品在TGA中表现出不同的失水特性,提示其晶体中结晶水的稳定状态不同,这种差异使其在加速实验中表现出色泽变化速率的不同;而注射用头孢噻肟钠晶体结晶度的差异是影响其稳定性的主要因素(未发表资料)。
上述结果提示,合理利用国家评价性抽验数据是发现国产制剂与参比制剂质量差异的有效途径。
3.2 根据杂质谱进行分析
2009年,中检院在国家评价性抽验中对不同成盐工艺的国产注射用阿奇霉素的杂质谱进行了考察,发现其中的主要杂质有阿奇霉素-N-氧化物、3'-去克拉定糖阿奇霉素、氮红霉素和阿奇霉素B;氮红霉素和阿奇霉素B为工艺杂质;阿奇霉素-N-氧化物和3'-去克拉定糖阿奇霉素分别为阿奇霉素的氧化和酸降解杂质,其中3'-去克拉定糖阿奇霉素主要在成盐过程中产生,并与成盐剂的种类密切相关,提示注射用阿奇霉素的成盐工艺是关键生产工艺;以3'-去克拉定糖阿奇霉素为指针,发现以柠檬酸(原研产品中的成盐剂)为代表的弱酸是理想的成盐剂,而盐酸等强酸不易作为注射用阿奇霉素的成盐剂[26]。
2014年,中检院在的国家评价性抽验中对阿莫西林克拉维酸钾片剂的杂质谱进行了分析,在17个厂家的152批样品中共发现有13个杂质,其中阿莫西林闭环二聚体和阿莫西林噻唑酸被认为是影响该品种关键质量属性的杂质;通过对其来源和降解途径的分析,认为生产中的关键工艺为制粒工艺,其中的干燥参数为关键工艺参数。此外,处方的合理性也是影响样品杂质产生的关键因素[27]。
2017年,中检院在的国家评价性抽验中对头孢拉定颗粒剂的杂质谱进行了分析,依据质量源于设计的理念,探寻其关键质量属性和关键工艺参数。在头孢拉定颗粒剂的特征杂质谱中有27个杂质,其中10个主要杂质的结构已经了确认,结合对不合格产品的杂质分析结果,确认生产过程中防止头孢拉定的氧化是关键工艺控制点,粒制剂过程中对制粒前温湿度的控制是工艺控制的关键[28]。
3.3 利于多变量数据分析方法
对某企业不同时间段生产的215批五水头孢唑林钠原料的28项质检项目包括:比旋度、吸收系数、pH值、水分、有关物质(杂质A、杂质B、杂质C、杂质F、杂质G、杂质H、杂质I、杂质J、杂质K、杂质L、杂质M、杂质N、单个未知杂质、总杂质)、聚合物、残留溶剂(N,N-二甲基乙酰胺、丙酮、异丙醇、二氯甲烷)、不溶性微粒(≥10μm不溶性微粒、≥25μm不溶性微粒)、松密度、紧密度、含量进行分析,所有产品均符合药品质量标准的规定。将检验数值(质量属性)经Z-score法标准化处理后,再利用one-way ANOVA分析,发现10项指标(比旋度、水分、杂质H、杂质I、杂质M、杂质N、总杂、丙酮、≥25μm不溶性微粒、紧密度)受批次变化(类别)的影响显著(P值<0.01),认为它们可能是影响产品质量的关键质量属性。通过对五水头孢唑林钠生产工艺的了解,上述10项指标分别与结晶过程、干燥过程和GMP厂房的洁净度控制环节相关联:结晶过程中的最适温度范围、持续时间等因素均可影响各种工艺杂质(杂质H、杂质I、杂质M和杂质N)在成品中的含量,进而影响产品的比旋度、总杂质含量等质量属性;结晶过程中有机溶剂的添加速率和温度等可影响晶体粒度的大小,进而影响成品的紧密度等质量属性。即在上述的10项指标中,7项(比旋度、杂质H、杂质I、杂质M、杂质N、总杂质、紧密度)与结晶工艺息息相关,产品间丙酮和水分含量的变异与干燥工艺相关,≥25μm不溶性微粒的数目可在一定程度上反映厂房的洁净程度。由此可知,结晶过程和干燥过程是五水头孢唑林钠原料药生产的关键工艺。利用这10项指标通过聚类分析,产品可被划分为不同的类;逐一分析这些指标对产品聚类结果的影响,发现比旋度可有效地反映出五水头孢唑林钠原料结晶工艺的变异;水分或丙酮的含量可反映干燥工艺对五水头孢唑林钠终产品的影响。即在上述可能影响产品质量的10个关键质量属性中,比旋度、水分/丙酮可用来表征五水头孢唑林钠生产过程一致性的最有效指针(未发表资料)。
4 抗生素注射剂参比制剂的选择
仿制药一致性评价过程中,需要以参比制剂实物标准,通过质量对比研究,保证仿制药品与参比制剂在临床中具有相同安全性与有效性。因此,参比制剂的选择直接影响仿制药一致性评价工作的质量。本文通过对3个典型抗生素注射剂的分析,探讨参比制剂选择过程中的关注点。
4.1 关注原研产品处方工艺的合理性
注射用头孢孟多酯钠的原研产品为头孢孟多酯钠与5%碳酸钠的混粉,1978年由美国礼来公司研发上市。贮存中注射用头孢孟多酯钠易水解产生头孢孟多,溶解时引起溶液的pH下降,可能导致溶液变浑浊。在产品中加入碳酸钠或氨基丁三醇可以改善溶液的浑浊[12]。国内的研究发现,含5%碳酸钠的注射用头孢孟多酯钠的溶液澄清度的不合格率明显降低,其总不良反应例数也远低于不含碳酸钠的注射用头孢孟多酯钠[13]。提示含5%碳酸钠的混粉产品更合理,即参比制剂应选择原研处方的注射用头孢孟多酯钠。
4.2 关注制药技术的发展方向
注射用头孢匹胺钠原研产品系1985年在日本上市的由日本住友制药公司与山之内制药公司联合开发的产品。因为当时没能找到适宜的成盐剂与头孢匹胺成盐,并获得头孢匹胺钠结晶,故选用成盐后直接冻干工艺生产;由于苯甲酸钠作为成盐反应的相转移催化剂无法从产品中去除,导致终产品为头孢匹胺钠与一定量的苯甲酸钠的混合物。伴随着科学技术的进步,目前已经寻找到适宜成盐剂,并能得到稳定的头孢匹胺钠晶体,使得溶媒结晶工艺成为生产头孢匹胺钠的主流工艺。相对于溶媒结晶工艺产品,冻干工艺的产品杂质含量偏高且稳定性较差,且由于含有一定量的苯甲酸钠,作为静脉给药的注射剂使其增加了不必要的安全性风险。近10年的不良反应监测结果揭示,溶媒结晶工艺产品的总不良反应例数远低于冻干工艺产品[29]。因此,参比制剂应选择溶媒结晶工艺的注射用头孢匹胺钠,以代表当前制药技术的发展方向。
4.3 关注产品的特殊性
注射用头孢唑林钠由日本藤泽药品株式会社于1971年开发上市。为保证产品的稳定性,原研最初采用结晶脱水工艺生产:即首先结晶先得到α-头孢唑林钠,再减压脱水得到脱水结晶性产品。结晶脱水工艺的生产成本较高,为降低生产成本,仿制药多采用冷冻干燥法生产无定型头孢唑林钠。无定型头孢唑林钠与胶塞的相容性较差,导致贮存过程中产品的澄清度极易不符合中国药典的规定[30]。伴随着对头孢唑林钠水合物特性的认知,通过完善结晶工艺,控制贮运条件等措施,头孢唑林钠水合物(α-头孢唑林钠、五水头孢唑林钠)产品分别在日本和国内上市。头孢唑林钠水合物不仅在成本上明显低于结晶脱水工艺产品,其制剂在杂质含量、与胶塞的相容性等方面均优于冻干法生产的无定型头孢唑林钠产品;头孢唑林钠水合物的严重不良反应的发生率也明显较无定型产品低[20];目前原研企业已经放弃结晶脱水工艺生产头孢唑林钠,而直接生产头孢唑林钠水合物(α-头孢唑林钠)。但头孢唑林钠水合物仅适合采用玻璃瓶包装。对以塑料膜为包材如双室袋等的新型制剂,由于包材的气密性较玻璃瓶差,较难保证贮运期间头孢唑林钠水合物的水分子不从晶格中逸出,因此该类制剂仍需以冷冻干燥法的无定型头孢唑林钠为原料进行生产。即结晶工艺生产的头孢唑林钠水合物与冷冻干燥法生产的无定型头孢唑林钠对特定的制剂类型均体现出一定的优势。对此类产品,是否需要针对不同的制剂类型选择不同的参比制剂值得思考。
5 结论
对抗生素注射剂的一致性评价应在对仿制药与原研药品安全性、有效性与生产一致性(manufacturing consistency)总体评估的基础上,企业通过对产品处方、工艺、质控水平的评估与提高,保证产品的临床疗效。多年来中检院组织的国家评价性抽验工作,已经积累了大量的国产仿制药质量信息,对其进行合理的分析与利用,有利于促进一致性评价工作的开展。适宜的参比制剂的选择直接影响一致性评价工作的质量。应以临床指标(安全、有效)为关键指针,选择处方、工艺合理、代表药品生产发展方向的主流产品作为参比制剂,且应考虑不同制剂的特殊性。
猜你喜欢
中西医结合心脑血管病杂志(2022年12期)2022-07-07 10:24:24
中国民间疗法(2021年4期)2021-06-09 09:20:10
中华养生保健(2020年2期)2020-11-16 00:49:20
药品评价(2020年13期)2020-10-14 03:50:40
工业设计(2020年6期)2020-07-30 14:05:39
中国生殖健康(2019年2期)2019-08-23 08:11:54
云南中医学院学报(2015年1期)2015-07-31 18:10:45
中国药业(2014年12期)2014-06-06 02:17:13
中国药业(2014年17期)2014-05-26 09:07:49
湖北科技学院学报(医学版)(2014年3期)2014-02-28 19:42:49
