
头孢拉定原料及制剂的聚合物杂质分析
2019-03-29 05:21:18李进张培培姚尚辰胡昌勤
中国抗生素杂志 2019年3期
李进 张培培 姚尚辰 胡昌勤
(中国食品药品检定研究院,北京 102629)
β-内酰胺类抗生素的高分子杂质容易诱发过敏反应,从而严重影响药物的安全性[1-4]。随着生产工艺水平的提高,药品生产过程控制日益严格,源于发酵工艺的外源性杂质,如残留蛋白、核酸 、多糖及其与抗生素的结合产物等[5],现在已得到有效控制,但是内源性的聚合物杂质可在药品原料与制剂生产、运输、存储过程中产生,目前仍是影响药物安全性的重要风险因素。为保证药品的安全性,对抗生素药物中的聚合物杂质应进行严格控制。
随着色谱分离技术的发展,聚合物的质控手段在不断进步,目前已报道的方法主要包括葡聚糖凝胶G10色谱法(Sephadex G10)、高效凝胶色谱法(HPSEC)、反相高效液相色谱法(RP-HPLC)、毛细管电泳法(HPCE)、液质联用(LC-MS)、柱切换技术、离子交换法等[6]。聚合物质控的理念逐渐由控制聚合物的总量向精准控制指针性聚合物杂质的方向发展[7]。在中国药典2015年版二部中,一共收录了26种头孢菌素类抗生素。其中12种采用葡聚糖G-10系统控制聚合物杂质;部分品种采用高效凝胶色谱法控制聚合物杂质,包括头孢米诺、头孢地嗪。
头孢拉定为第一代半合成头孢菌素,国家基本药物,具有耐酸、耐酶、吸收好、血药浓度高等优点,临床主要用于治疗呼吸道感染、泌尿生殖道感染及皮肤软组织感染等。中国药典中头孢拉定的聚合物的质控方法为葡聚糖凝胶G10色谱法[8],美国药典41版[9]未收载该品种,欧洲药典(EP)9.0版[10]中未设定头孢拉定聚合物控制项。
传统葡聚糖凝胶G-10系统存在柱效较低、操作不方便、分离时间长、分离度差等不足,少数头孢菌素类抗生素采用了高效凝胶色谱法控制聚合物杂质的总量。国外药典虽然未设定专门的聚合物检查项,但是在部分具体品种的有关物质项下,如阿莫西林、头孢噻肟钠等采用RP-HPLC方法控制其中的二聚体、三聚体等,但是该类方法能否检出更高聚合态的聚合物杂质缺乏方法专属性研究。在前期研究中,已对青霉素类抗感染药物聚合物杂质做过研究工作,证明采用中国药典2015年版的RP-HPLC有关物质测定方法可对指针性聚合物杂质进行精准控制[11-12]。本文首先采用高效凝胶色谱法(HPSEC)和柱切换-LC/MS法对头孢拉定聚合物进行分离及初步定性分析;然后以EP9.0头孢拉定有关物质分析方法为基础,优化建立了头孢拉定的聚合物分析方法,并对该方法进行了方法学验证,进而为头孢菌素类抗生素聚合物的精准控制提供了可借鉴的参考。
1 材料与方法
1.1 仪器
二维色谱系统为Summit 100型,包括P680型双三元低压梯度泵、ACI-100型自动进样器、Tcc-100型柱温箱及PDA-100型二极管阵列检测器组成,工作站为Chromeleon 6.8版(美国Dionex公司);柱切换LC-MS系统由资生堂HPLC色谱系统(包括Nano Space S1-2二元高压梯度泵、自动进样器、柱温箱、切换阀和PDA检测器 )和Qtrap 3200型MS/MS质谱仪(美国ABsciex公司)组成,工作站为Analyst 1.6版;Buchi旋转蒸发仪R-215V。
1.2 样品与试剂
头孢拉定原料(批号:130588-201202)由中国食品药品检定研究院提供。
乙腈(色谱纯)购自美国Fisher公司,其他化学试剂(分析纯),均购自国药集团化学试剂公司,水为实验室自制双蒸水。
1.3 高效凝胶排阻色谱(HPSEC)法
色谱柱TSK-gel G2000SWxl(填料:刚性、球形、亲水硅胶;7.8mm×30cm,5μm);流动相:A相:磷酸盐缓冲液(pH7.0)[0.01mol/L磷酸氢二钠溶液-0.01mol/L磷酸二氢钠溶液(61:39)],B相:乙腈,A:B=85:15,等度洗脱;流速:0.7mL/min;检测波长:233nm;柱温:30℃;进样量:20μL。稀释溶剂:水;样品浓度:10.0mg/mL。用于HPSEC法分析聚合物杂质。
1.4 RP-HPLC法
色谱柱:Agilent ZORBAXS-BC18(4.6mm×150mm,5μm),PN:883975-902,SN:USCM040539, LN:B14172;流动相:A相:2.72g/L磷酸氢二钾溶液(用磷酸溶液调节pH值=3.0);B相:甲醇,梯度洗脱0~2.5min,A相由99.5%降低至97.0%,2.5~11.0min降低A相至75%,11.0~13.0min降低A相为60%,13.0~16.0min维持A相为60%,16.0~19.0min降低A相至20%,19.0~30.0min维持A相为20%,30.0~30.1min升高A相至99.5%,30.1~35.0min维持A相为99.5%;流速:1.0mL/min;柱温:30℃;检测波长:220nm。用于分析头孢拉定原料及制剂中的指针性聚合物杂质。
1.5 二维色谱(2D-HPLC)法
色谱系统I同“1.3”项高效凝胶色谱法;色谱系统II同“1.4”项RP-HPLC优化法。切换阀的连接方式如图1所示,切换程序:①将切换阀设为A位置,采用右泵,以TSK凝胶色谱系统(色谱系统I)对供试品进行分离,同时采用色谱系统II对RP C18色谱柱进行平衡。②当采用色谱系统I分离的目标杂质出峰后,将切换阀设为B位置。③采用色谱系统II将柱后定量环中的目标杂质洗脱至第二根色谱柱中,并采用色谱系统II进行分离分析。用于对HPSEC色谱系统分离的聚合物杂质在RP-HPLC色谱系统中进行定位。
1.6 柱切换-LC/MS-I法
色谱系统III:同“1.3”项色谱系统,进样体积增加至50μL,用于杂质的分离。
色谱系统IV:色谱柱:KromasiL 100-5,C18(SN:E68355,4.6mm×250mm,5μm)。流动相:A相:含有1%甲酸的水溶液;B相:含有1%甲酸的乙腈溶液;梯度洗脱:0~(tR+5min)(tR为目标杂质在色谱系统III的保留时间)维持100%的A相(脱盐处理),(tR+5)~(tR+20)min 将A相降低至10%,(tR+20)~(tR+25)min维持A相为10%,(tR+25)min ~(tR+26)min升高A相至100%,(tR+26)~(tR+40)min,维持A相100%。柱温:室温;流速:0.5mL/min。切换阀:六通阀A和B;切换用定量环体积:500μL;用于对高效凝胶色谱系统分离的聚合物杂质进行脱盐处理、质谱定性研究。
1.7 柱切换-LC/MS-II法
色谱系统V:同“1.4”项色谱系统,进样体积增加至50μL,用于杂质的分离。
色谱系统VI:同色谱系统IV,其中tR为目标杂质在色谱系统V的保留时间。用于对RP-HPLC色谱系统分离的聚合物杂质进行脱盐处理、质谱定性研究。
1.8 质谱条件(“1.6”和“1.7”项的质谱方法)
+EMS和+EPI扫描范围:m/z200~1700;气帘气:20L/min;离子源电压:+5000V;离子源温度:400.00℃;气路1:65L/min;气路2:60L/min;碰撞气强度:强;解簇电压:+70.0V;入口电压:+10.0V;碰撞室入口电压:+10.00V;碰撞能:+10.00V:碰撞室出口电压:+0.00V。
1.9 溶液配制
头孢拉定强制降解物:取头孢拉定供试品约500mg,置于玻璃试管中,加2mL氯仿,再加0.4mL三乙胺,振摇3min后,密封保存。室温静置24h,待完全溶解后,用旋转蒸发仪在40℃水浴中蒸干溶剂,密封避光保存,作为头孢拉定强制降解物。

图1 2D-HPLC中10孔切换阀示意图Fig.1 Schemes of the 10-hole switching valve in 2D-HPLC
头孢拉定强制降解溶液:称取上述头孢拉定强制降解物适量,加水溶解并定量稀释成约含有10mg/mL的溶液,摇匀备用,作为头孢拉定强制降解溶液,备用。
方法学验证用溶液:精密称取头孢拉定强制降解物适量,加水溶解并定量稀释成浓度约为5mg/mL的溶液,作为方法学验证溶液。
2 结果与讨论
2.1 高效凝胶色谱法(HPSEC)分析头孢拉定聚合物
本研究首先参照中国药典2015年版二部中头孢地嗪有关物质II的方法[13],建立了TSK G2000 SWxL高效凝胶排阻色谱法(HPSEC),对头孢拉定强制降解溶液进行初步分离。结果显示,在头孢拉定主峰前检出若干个弱保留值杂质HPSEC-1~5等;其中HPSEC-1、2、3未能完全分离,参见图2b;故在中国药典方法基础上,改变流动相中强溶剂的比例,对HPSEC法进行方法优化,结果如图2a、c和d所示。当流动相A:B=85:15时,弱保留值杂质HPSEC-1与HPSEC-2、HPSEC-3达到基线分离,但是各弱保留值杂质色谱峰拖尾严重,部分色谱峰还存在肩峰,分离效果较差。
2.2 柱切换-LC/MSn法鉴别头孢拉定聚合物的化学结构
以头孢拉定强制降解溶液为供试品,采用“1.3”项的优化后高效凝胶色谱法,在头孢拉定主峰前分离得到弱保留值杂质HPSEC-1~5,其中包括聚合物杂质和小分子杂质,然后采用柱切换-LC/MS-I法(参见“1.6”项)法鉴别各组分的化学结构。
2.2.1 弱保留值杂质HPSEC-1
柱切换-LC/MS结果表明HPSEC-1中包含多种小分子杂质,不是聚合物杂质。
2.2.2 弱保留值杂质HPSEC-2和3
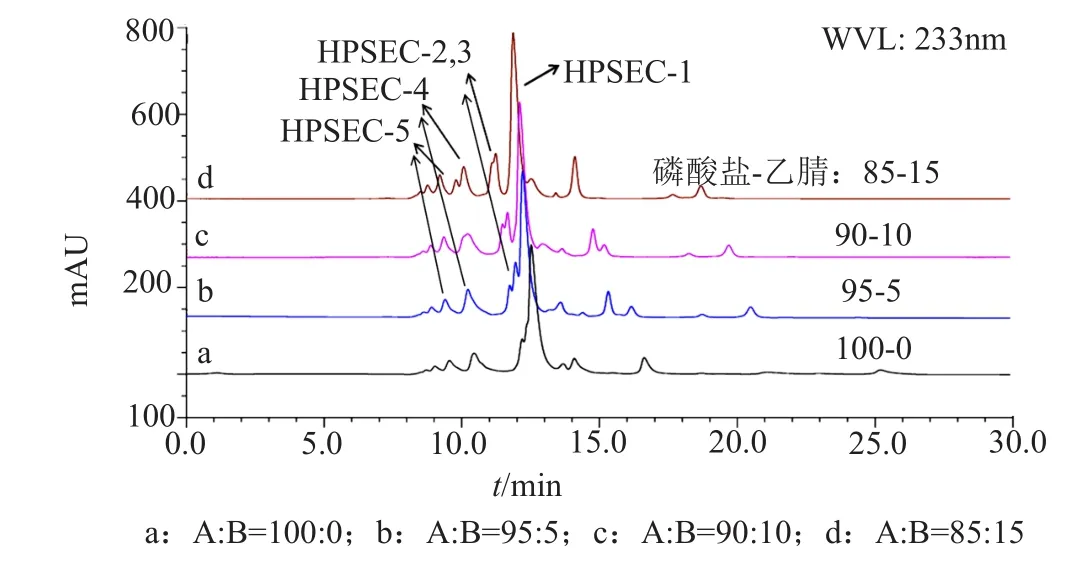
图2 HPSEC法分析头孢拉定强制降解溶液的典型色谱图Fig.2 Typical chromatograms of cefradine stress degradation solution by HPSEC
弱保留值杂质HPSEC-2和3的TIC图、+EMS图及+EPI@m/z699.4[M+H]+的二级质谱如图3a所示。+EMS图均显示m/z699.5、m/z721.3的准分子离子峰,分别为[M+H]+、[M+Na]+峰,推测其分子量为698.2,是头孢拉定分子量的2倍,提示其为头孢拉定二聚体,形成机理推测为一分子头孢拉定7位侧链的氨基,与另一分子头孢拉定母核四元内酰胺环的羰基反应,形成酰胺键从而聚合形成二聚体。HPSEC-1和HPSEC-2的一级和二级质谱图基本一致,提示二者互为同分异构体。异构化的反应机理推测为头孢拉定在碱性条件下发生开环反应时6位和7位的手性碳构型发生改变,从而发生异构化,形成头孢拉定异构体,理论上存在4种异构体。头孢拉定二聚体及其异构体的结构参见图4a。质谱分析结果证明弱保留值杂质HPSEC-2和HPSEC-3为头孢拉定二聚体。
2.2.3 弱保留值杂质HPSEC-4
弱保留值杂质HPSEC-4的TIC图、+EMS图及+EPI@m/z1048.6[M+H]+的二级质谱如图3b所示。+EMS图存在m/z1048.6、m/z1070.4的准分子离子峰,分别为[M+H]+、[M+Na]+峰,提示其分子量为1047.6,为头孢拉定分子量的3倍,推测为头孢拉定三聚体,理论上也应存在多种异构体,结构见图4b。
2.2.4 弱保留值杂质HPSEC-5
弱保留值杂质HPSEC-5的TIC图、+EMS质谱数据如图3c所示。+EMS图显示m/z 1397.8的准分子离子峰,为[M+H]+峰,提示其分子量为1396.8,为头孢拉定分子量的4倍,推测为头孢拉定四聚体,理论上推测也存在多种异构体。化学结构参见图4c。
2.3 RP-HPLC法分析头孢拉定的聚合物杂质
高效凝胶色谱法分析头孢拉定聚合物杂质的方法专属性较差,因此我们尝试建立了RP-HPLC法,并采用二维色谱法和柱切换-LC/MS法对RP-HPLC的专属性进行研究。
2.3.1 建立RP-HPLC法分离聚合物杂质
在EP9.0头孢拉定有关物质测定方法的基础上,通过增大有机相的比例,延长洗脱时间的方法,优化建立了聚合物分析的RP-HPLC法,具体方法参见“1.4”项,RP-HPLC法分析头孢拉定强制降解溶液的典型色谱图如图5所示。
2.3.2 二维色谱法验证RP-HPLC法分离聚合物杂质的专属性

图3 HPSEC-2~5的+EMS(1)与+EPI(2)质谱图Fig.3 +EMS(1)and +EPI(2)mass spectra of impurities HPSEC-2~5

图4 强制降解溶液中聚合物杂质的结构式Fig.4 Structures of polymer impurities in stress degradation solution
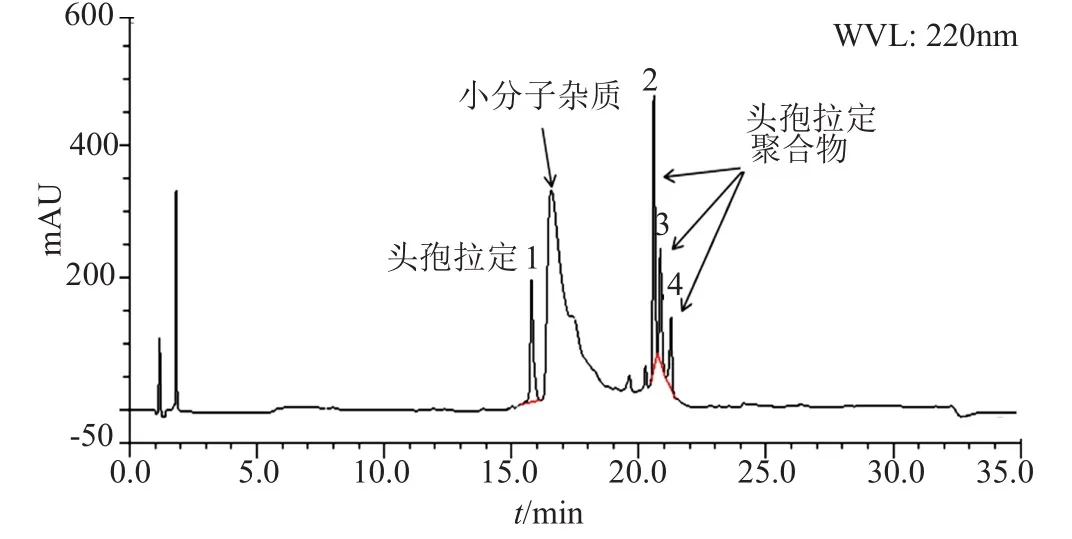
图5 头孢拉定强制降解溶液的典型RP-HPLC图Fig.5 Typical chromatograms of cefradine stress degradation solution by RP-HPLC
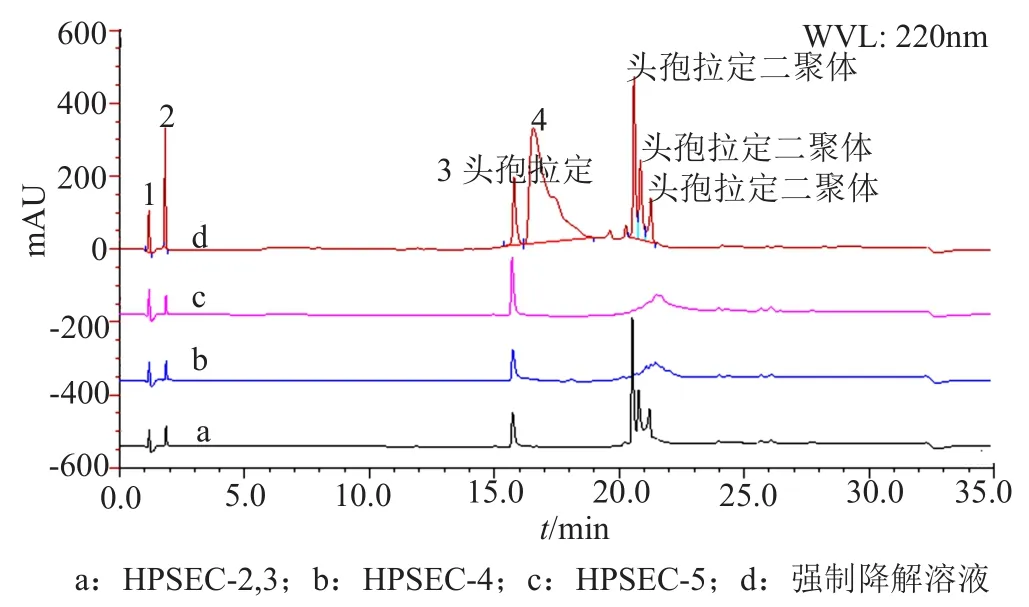
图6 杂质HPSEC-2~5与强制降解溶液的典型二维色谱图Fig.6 Typical two-dimensional chromatograms of impurityHPSEC-2~5 and stress degradation solution
为了考察RP-HPLC法分离聚合物杂质的专属性,本文采用二维色谱法(“1.5”项),将高效凝胶色谱法分离的聚合物杂质HPSEC-2~4,转移至RPHPLC系统中进行分析,归属各种聚合物杂质的出峰位置,如图6所示。结果显示头孢拉定二聚体、三聚体、四聚体在RP-HPLC系统中的出峰时间均为20.0~22.0min,其中HPSEC-2色谱峰在反相系统中分离为3个色谱峰,HPSEC-3和HPSEC-4在反相系统中均分离为多重色谱峰,这与前期推断头孢拉定聚合物存在多种异构体的结论相吻合。因此,头孢拉定强制降解物的RP-HPLC典型图谱中20.0~22.0min的一组色谱峰可以初步归属为头孢拉定聚合物杂质,聚合物杂质出峰集中,且不受其他降解的小分子杂质干扰,初步说明建立的RP-HPLC分析头孢拉定聚合物杂质的专属性较好。
2.3.3 柱切换-LC/MS法验证RP-HPLC法分离聚合物杂质的专属性
为了确认RP-HPLC法分析聚合物杂质的专属性,本文采用柱切换-LC/MSn-II法,将RP-HPLC色谱系统中20.0~22.0min区间的3个主要杂质峰Imp-1~3分别进行质谱分析,结果如图7所示。+EMS质谱图显示3者均存在m/z699、m/z721、m/z737的准分子离子峰,分别为 [M+H]+、[M+Na]+、[M+k]+峰,推测其分子量均为698,与头孢拉定二聚体的分子量一致,证明这3个主要色谱峰均为头孢拉定二聚体,三者互为同分异构体,与二维色谱法的分析结果相一致。
2.3.4 RP-HPLC法分离聚合物杂质的方法学验证
为了保证建立的方法灵敏、准确、耐用,本研究对RP-HPLC法进行了方法学验证。
(1)专属性实验:参见“2.3.2”和“2.3.3”项。
(2)检测限与定量限:精密量取方法学验证溶液5μL,用水进行倍比稀释,得到不同浓度的系列溶液,注入液相色谱仪。以基线噪音的10倍为指标,得到方法的最低定量限;以基线噪音的3倍为指标,得到方法的最低检测限。结果表明,最低定量限为500μg;最低检测限为150μg。
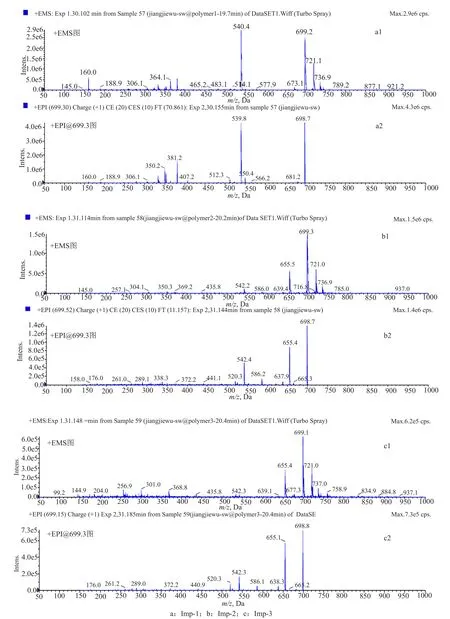
图7 强制降解溶液在RP-HPLC系统中3个杂质主峰的典型+EMS(1)和+EPI(2)质谱图Fig.7 Typical +EMS (1)and +EPI (2)mass spectrograms of 3 impurity peaks in stress degradation solution separated by RP-HPLC
(3)重复性实验:精密量取方法学验证溶液5μL,注入液相色谱仪,连续进样3针,计算3个二聚体杂质的峰面积之和,经计算,3次重复进样的RSD为0.12%。
(4)耐用性实验:精密量取方法学验证溶液5μL,注入液相色谱仪,考察在不同柱温、流动相pH值、和不同色谱柱条件下,聚合物杂质的分离情况。如图8所示。结果表明当柱温、流动相pH值、色谱柱型号发生轻微改变时,RP-HPLC法均可有效分离样品中的头孢拉定聚合物杂质,头孢拉定聚合物杂质的保留时间维持在20.0~22.0min范围内。
3 结论
本文综合运用高效凝胶色谱法(HPSEC)、二维色谱法(2D-HPLC)、柱切换-LC/MSn法等现代色谱分析技术,证明了基于TSK2000 SWxL型高效凝胶柱系统的HPSEC法分析头孢拉定原料及制剂的聚合物杂质的方法专属性差,新建立的RP-HPLC法分析头孢拉定聚合物杂质灵敏度高、耐用性好、专属性强,适用于头孢拉定原料及制剂的聚合物质控;头孢拉定强制降解溶液可作为头孢拉定聚合物分析的系统适用性溶液。
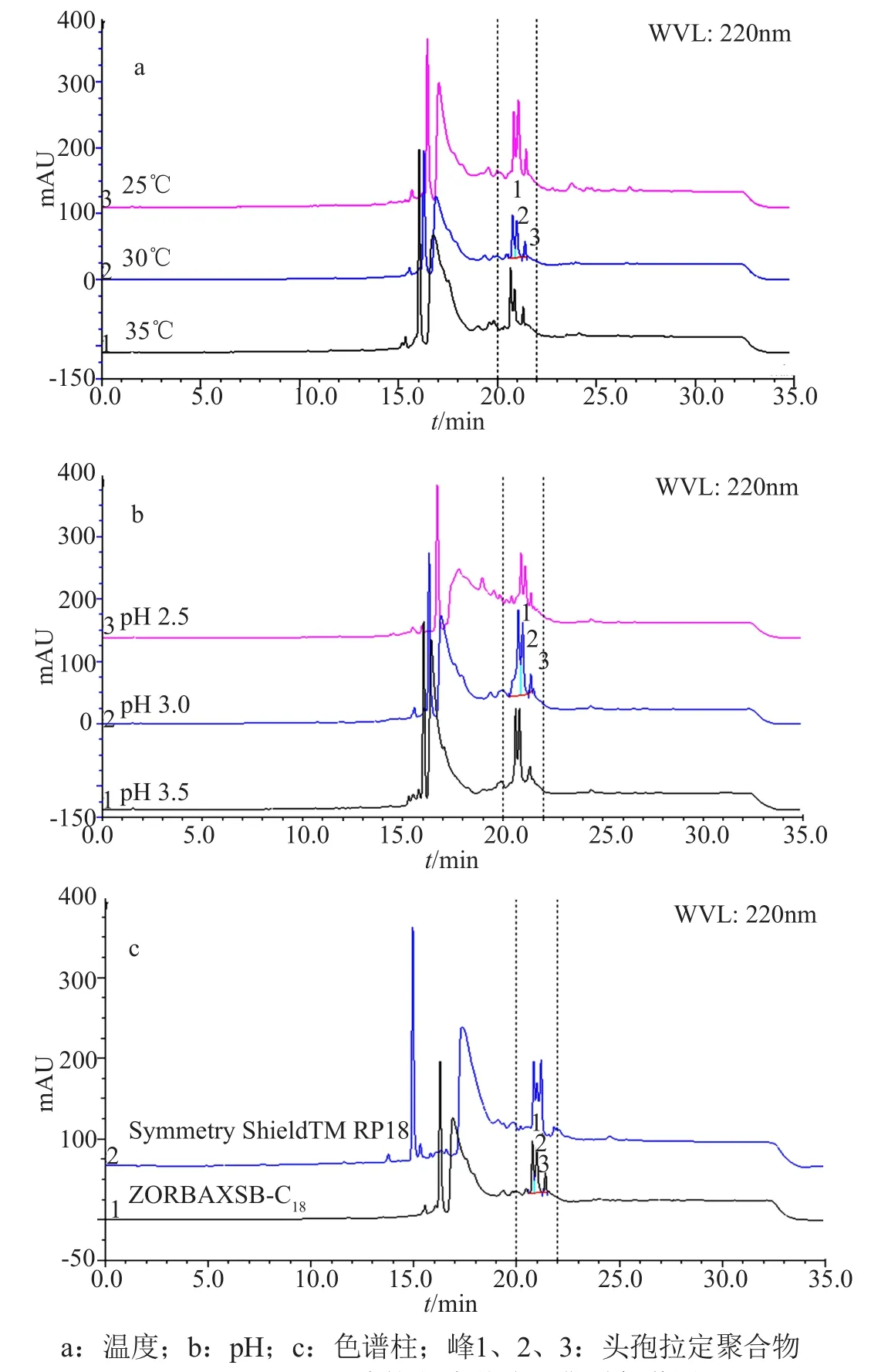
图8 RP-HPLC法的方法学验证典型色谱图Fig.8 Typical RP-HPLC chromatograms for method validations
猜你喜欢
环境保护与循环经济(2021年7期)2021-11-02 08:10:52
食品安全导刊(2021年20期)2021-08-30 06:39:22
艺海(2018年2期)2018-08-29 00:04:46
科学与财富(2017年32期)2017-12-20 22:13:54
科学与财富(2017年29期)2017-12-20 20:59:54
学术论坛(2016年5期)2016-05-17 05:44:43
乐活老年(2016年10期)2016-02-28 09:30:32
河北工业科技(2015年4期)2015-02-27 13:15:36
中国药业(2014年12期)2014-06-06 02:17:26
中国合理用药探索(2014年11期)2014-03-11 20:30:22
