
p53 mutation regulates PKD genes and results in co-occurrence of PKD and tumorigenesis
2019-03-23 05:16HailiLiYongjinZhangJuhuaDanRuoyuZhouCuiLiRongLiXiaomingWuSanjayKumarSinghJeffreyChangJulunYangYingLuo1
Cancer Biology & Medicine 2019年1期
Haili Li, Yongjin Zhang, Juhua Dan, Ruoyu Zhou, Cui Li, Rong Li, Xiaoming Wu, Sanjay Kumar Singh,Jeffrey T. Chang, Julun Yang, Ying Luo1,
1Faculty of Environmental Science and Engineering, Kunming University of Science and Technology, Kunming 650500, China;2Laboratory of Molecular Genetics of Aging &Tumor, Kunming University of Science and Technology, Kunming 650500,China; 3Division of Nephrology, The First People's Hospital of Yunnan Province, Kunming 650032, China; 4Department of Cancer Systems Imaging, The University of Texas MD Anderson Cancer Center, Houston 77030, TX, USA; 5Department of Integrative Biology and Pharmacology, University of Texas Health Science Center at Houston, Houston 77030, TX, USA;6Department of Pathology, Kunming General Hospital, Kunming 650032, China
ABSTRACT Objective:Polycystic kidney disease (PKD) is the major cause of kidney failure and mortality in humans. It has always been suspected that the development of cystic kidney disease shares features with tumorigenesis, although the evidence is unclear.Methods:We crossed p53 mutant mice (p53N236S, p53S) with Werner syndrome mice and analyzed the pathological phenotypes.The RNA-seq, ssGSEA analysis, and real-time PCR were performed to dissect the gene signatures involved in the development of disease phenotypes.Results:We found enlarged kidneys with fluid-filled cysts in offspring mice with a genotype of G3mTerc-/-WRN-/-p53S/S (G3TM).Pathology analysis confirmed the occurrence of PKD, and it was highly correlated with the incidence of tumorigenesis. RNA-seq data revealed the gene signatures involved in PKD development, and demonstrated that PKD and tumorigenesis shared common pathways, including complement pathways, lipid metabolism, mitochondria energy homeostasis and others. Interestingly, this G3TM PKD and the classical PKD1/2 deficient PKD shared common pathways, possibly because the mutant p53S could regulate the expression levels of PKD1/2, Pkhd1, and Hnf1b.Conclusions:We established a dual mouse model for PKD and tumorigenesis derived from abnormal cellular proliferation and telomere dysfunction. The innovative point of our study is to report PKD occurring in conjunction with tumorigenesis. The gene signatures revealed might shed new light on the pathogenesis of PKD, and provide new molecular biomarkers for clinical diagnosis and prognosis.
KEYWORDS p53 mutation; telomere dysfunction; polycystic kidney disease; tumorigenesis
Introduction
Polycystic kidney disease (PKD) is a disease where enlarged kidneys develop characteristic fluid-filled cysts. Cysts in the liver or pancreas, cerebral aneurysms, abnormal cardiac development, and hypertension are also frequently found in PKD patients. Genetic studies have shown that approximately 80% of autosomal dominant PKD (ADPKD)is caused by mutations in the PKD1 gene (encoding polycystin-1, PC1), and about 20% of ADPKD was due to mutations in PKD2 gene (encoding polycystin-2, PC2). It has been extensively shown that PC1/2 act as the key regulators for calcium homeostasis, and the dysfunction of PC1/2 might play an essential role in calcium imbalance and cAMP signaling, resulting in the development of PKD phenotypes1,2. Increasing evidence suggests that PC1/2 proteins might interact with key regulators in cell cycle regulation, especially in cell proliferation and secretionrelated signaling pathways1. PKD1 has been found to play a role in preventing immortalized proliferation of renal cells through p53 and JNK, suggesting a novel link between PKD1 and p533. It has also been found that the tumor suppressor protein p53 participates in a negative feedback loop to regulate PKD1 gene expression, thus preventing renal cysts formation4. Interestingly, another study has shown that Mekk1 acts as a co-repressor with p53 to downregulate PKD1 transcription. This PKD1 repression could be promoted by stress stimuli, suggesting that abnormally elevated stress responses might directly downregulate the PKD1 gene,possibly causing haploinsufficiency and cyst formation5. In an endothelial cell-culture system, elevated expression of mechanosensory polycystins in human carotid atherosclerotic plaques is associated with p53 activation and disease severity6. At the animal level, Bcl2 knockout mice manifested PKD and PKD phenotypes that could not have been rescued by p53 deficiency7,8. The mutant p53 protein,especially the missense point mutation, is the major form of p53 deficiency in human disease. It promotes the progress of disease by both loss and gain of function9. However, no evidence has been found to connect mutant p53 with the progress of PKD.
Werner syndrome (WS) protein is a member of the RecQ helicase family implicated in the maintenance of genome stability. WRN plays an essential role in telomere DNA replication, and WRN defects cause human pathologies linked to cancer predisposition and premature aging, such as WS10-12. By masking the chromosome ends from the DNA repair machinery through repression of the ATM/ATR signaling pathways, telomere DNA has a crucial function in DNA damage response (DDR). Telomere DNA is elongated by telomerase and protected by the protein complex shelterin, which regulates telomere length and protects telomeres from activating DDR13.
The mouse model of WS is established by double knockout of WRN and the RNA component of telomerase. The late generation (G4-6) of WS mice with both telomerase and WRN deficiency (mTR-/-WRN-/-) exhibited the clinical features observed in WS patients14-16. Our previous study has shown that ALT tumorigenic cell lines derived from senescent WS MEFs gained the same point mutation in tumor suppressor gene Trp53, encoding a mutant p53 protein known as p53N236S (p53S hereafter). The p53S/Smice manifested highly invasive lymphomas and metastatic sarcomas with dramatically increased double minute chromosomes17.
We introduced this p53S mutation back into WS mice to study the intrinsic role of p53S in modulating WS symptoms,by crossing mice carrying p53S mutation with WS mice.Surprisingly, we found that the offspring of p53S and WS mice (mTR-/-WRN-/-p53S/S) manifested both PKD and tumor phenotypes. Here we report the phenotypes of this novel mouse model. By RNA-seq and ssGSEA analysis, we have identified the gene signatures and pathways that connect mutant p53 and telomere dysfunction with the development of PKD.
Materials and methods
Mice
Transgenic p53S mice and WS (mTR-/-WRN-/-) mice were bred to generate mTR-/-WRN-/-p53S/Smice. We crossed mice carrying p53S mutation (p53S/S) with WS mice (mTR-/-WRN-/-)and obtained the first generation of mice with telomerase knockout, WRN knockout, and p53S mutation (G1 mTR-/-WRN-/-p53S/S), referred to as G1 triple mutation (G1TM).The mice were then bred generation-by-generation to obtain G2 and G3 TM mice. The telomerase knockout and WRN knockout mice (double mutation, DM) and wild type (WT)mice were used as control. All experiments were carried out with the approval of the Kunming University of Science and Technology and Use Committee (Approval ID: M2015-011)in accordance with the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care.
MEF cells
The MEF cells with different genotypes were harvested in 13.5 days and cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) at 37 °C with 5% CO2and 3% O2. To maintain their original characteristics, only the early passages (≤ passage 5) of MEF cells were used for experiments.
Pathology analysis
Mouse kidney samples were fixed in 4% neutral buffered formalin for 6 hours, then alcohol-dehydrated and paraffinembedded. The paraffin-embedded tissue blocks were sectioned into 4 μm slices for later experiments. For hematoxylin-eosin (HE) staining, the tissue sections were deparaffinized and rehydrated, and H&E staining was applied. The H&E stained slides were observed via microscopy and the histological changes and kidney lesions were evaluated by pathologists.
RNA-seq and gene expression signature analysis
Cell or tissue (sarcoma and cystic kidney) samples were collected and sent for commercia RNA-seq service(Novogene, China). Briefly, the total RNA was extracted and enriched by oligo-dT labeled magnetic beads, and used to construct a library for RNA-seq. The sequenced reads (raw reads) were evaluated for quality control. The adapters and low quality reads were filtered to obtain clean reads. The clean data were then aligned with the reference mouse genome by TopHat2. The RNA-seq counts were annotated and the FPKM file was generated for bioinformatic analysis.The Bioinformatics ExperT SYstem (BETSY) was applied to automate the development of workflows18. The single sample gene set enrichment analysis (ssGSEA)19was applied to analyze the RNA-seq data. Hallmark (designed for welldefined biological states and processes), C2 (BIOCARTA,KEGG, REACTOME, etc.), and C5 (GO) gene sets from the Molecular Signatures Database20were used for ssGSEA analysis. The heat maps were plotted with BETSY by centering with mean but without hierarchical clustering. The common pathways between cystic kidneys and tumors were ranked and plotted based on their ssGSEA scores.
Ingenuity pathway analysis
The essential genes involved in PKD development were selected according to the literature1,21. The fold change in their expression between G3TM and G3DM was calculated from RNA-seq data. After applying the cutoff (2 ×) for gene expression fold change, the remaining genes and their fold changes, and P values were imported to Ingenuity Pathway Analysis (IPA) software. The knowledge base of IPA were used to draw their expression regulation and interaction network. The network with largest numbers of genes is included, such as developmental disorders, immunological diseases, inflammatory diseases, inflammatory response, and renal and urological disease.
Quantitative real-time PCR analysis
RNA was isolated from cell or tissue samples, and cDNA was synthesized by reverse transcription. Real-time PCR was performed on an ABI Prism 7300 sequence detection system with SYBR-Green PCR master mix according to the manufacturer's instructions (Applied Biosystems, CA). The primers used are as follows:
PKD1, forward primer: 5'-CCCTCTCGGAGCAGAA TCAAT-3', reverse primer: 5'-GTGTTGAGCTAATGGGC AGG-3';
PKD2, forward primer: 5'-GGGGAACAAGACTCATG GAAG-3', reverse primer: 5'-GCCGTAGGTCAAGATGC ACAA-3';
Pkhd1, forward primer:5'-GGGAGGTCGATGGTGCA TAAG-3', reverse primer: 5'-GATGTCCGTTCTTCCCCC AAG-3';
Hnf1b, forward primer: 5'-AGGGAGGTGGTCGATG TCA-3', reverse primer: 5'-TCTGGACTGTCTGGTTGA ACT-3';
C2, forward primer: 5'-CGGTGGTAATTTCACCCTCAG-3', reverse primer: 5'-GGTGTGATGTGAGCTAGACCT-3';
C5, forward primer: 5'-GAACAAACCTACGTCATTTCA GC-3', reverse primer 5'-GTCAACAGTGCCGCGTTTT-3';
Pgc1a, forward primer: 5'-TATGGAGTGACATAGAGTGT GCT-3', reverse primer: 5'-CCACTTCAATCCACCCAGAAA G-3';
Tfam, forward primer: 5'-ATTCCGAAGTGTTTTTC CAGCA-3', reverse primer: 5'-TCTGAAAGTTTTGCATCTG GGT-3';
Wnt1, forward primer: 5'-GGTTTCTACTACGTTGCTA CTGG-3', reverse primer: 5'-GGAATCCGTCAACAGGTT CGT-3';
Ctnnb1, forward primer: 5'-ATGGAGCCGGACAGAAA AGC-3', reverse primer: 5'-CTTGCCACTCAGGGAAG GA-3';
Srebf1, forward primer: 5'-GATGTGCGAACTGGACA CAG-3', reverse primer: 5'-CATAGGGGGCGTCAAAC AG-3';
Srebf2, forward primer: 5'-GCAGCAACGGGACCAT TCT-3', reverse primer: 5'-CCCCATGACTAAGTCCTTCAA CT-3';
β-actin, forward primer: 5'-AGAGGGAAATCGTGCG TGAC-3', reverse primer: 5'-CAATAGTGATGACCTGGCC GT-3'.
Results
Generation of a mouse model manifesting PKD phenotypes
We crossed mice carrying p53S mutation with WS mice and obtained the first generation of mice with telomerase, WRN knockout, and p53S mutations (G1mTR-/-WRN-/-p53S/S),referred to as G1 triple mutation (G1TM). The mice were then bred generation-by-generation to obtain G2 and G3 TM mice (Figure 1A and 1B).
As expected, we observed the incidence of sarcomas when telomere length was shortened to a certain level, which occurred in G3TM (G3mTR-/-WRN-/-p53S/S) mice (Figure 1C). The affected mice were sacrificed and anatomical analysis showed that the mice also manifested unilateral or bilateral enlarged kidneys with multiple fluid-filled cysts(Figure 1D). Thus, surprisingly, PKD phenotypes were found in G3TM mice at around 4 months old.
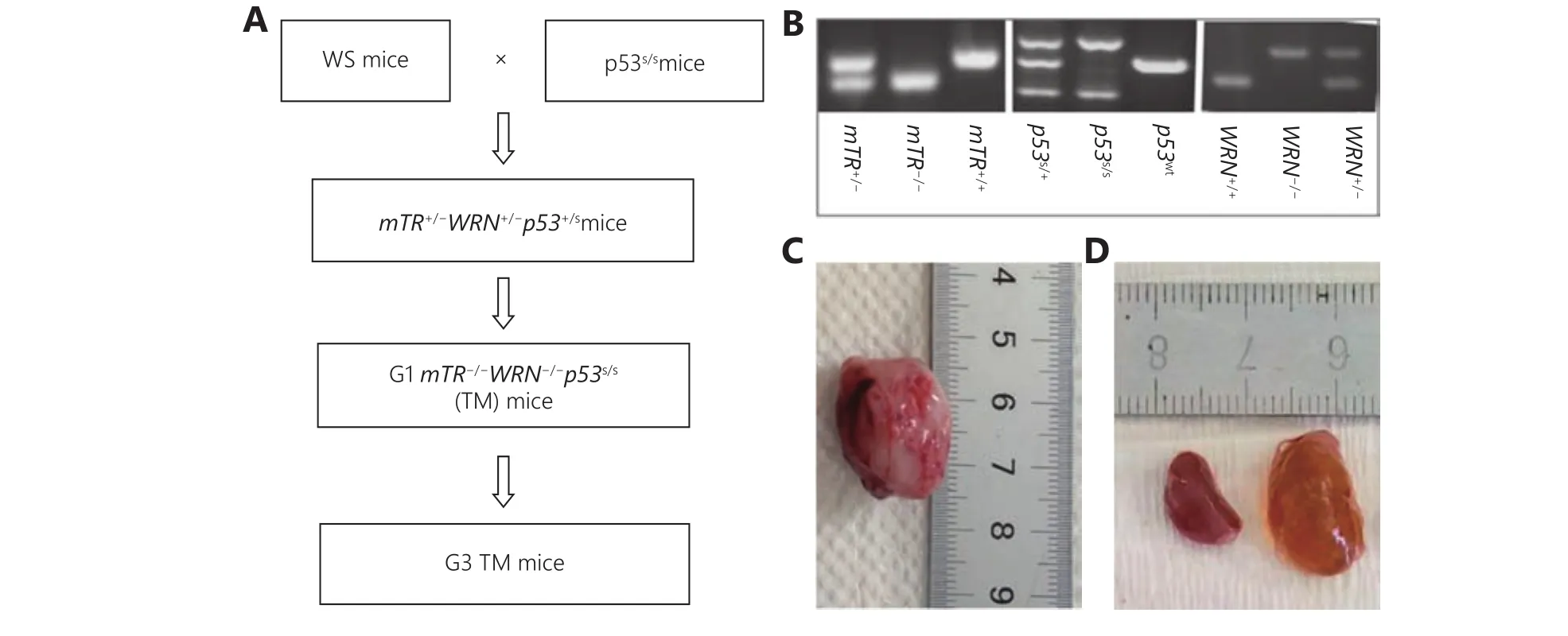
Figure 1 Generation of a mouse model manifesting PKD. (A) The breeding strategy for generating G3TM (G3mTR-/-WRN-/-p53S/S). Mice carrying the p53S mutation were crossed with WS mice and G1TM were obtained (G1mTR-/-WRN-/-p53S/S). The mice were then bred generation-by-generation to obtain G2 and G3 TM mice. (B) Genotyping of mice carrying mTR, WRN and p53S mutations. (C) Incidence of sarcoma in a G3TM mouse. (D) Bilateral enlarged kidneys with multiple fluid-filled cysts in a G3TM mouse.
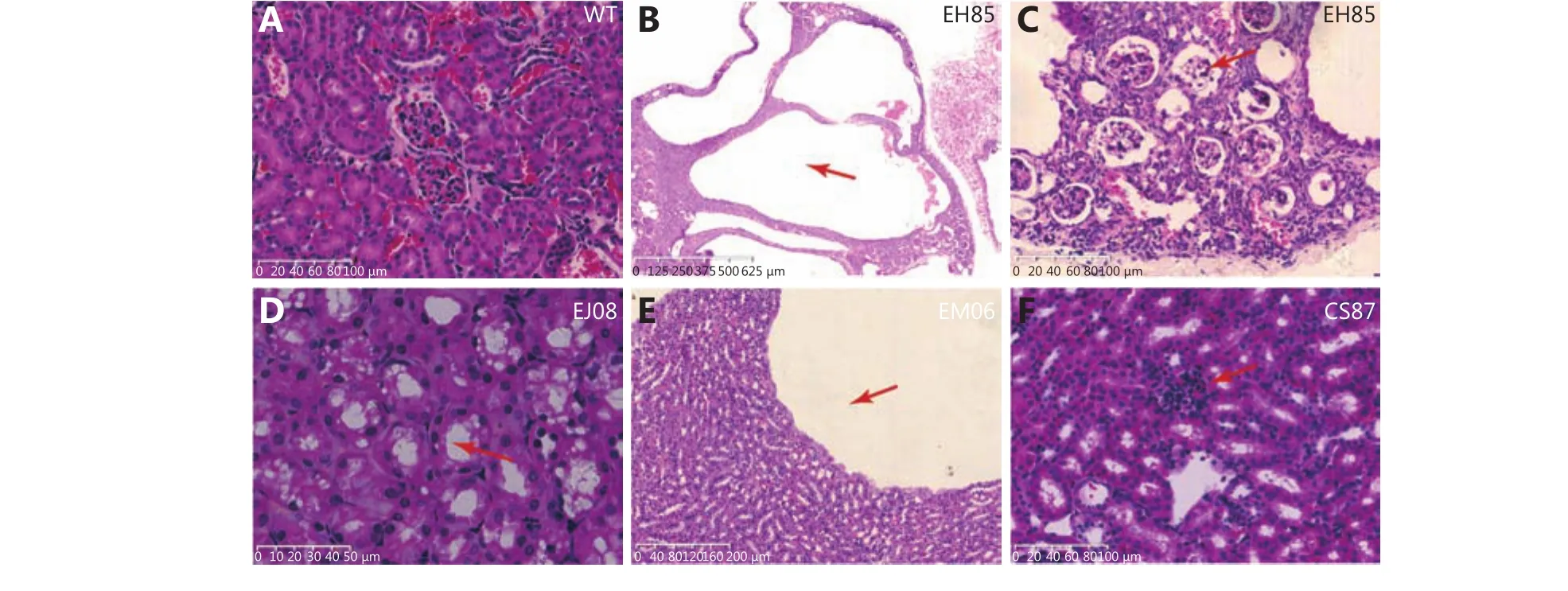
Figure 2 Hematoxylin and eosin staining of kidney sections from mice with PKD phenotype. (A) The normal morphology of a kidney from a wild type mouse. (B) An end stage cystic kidney from a G3TM (G3mTR-/-WRN-/-p53S/S) mouse (ID number: EH85). The normal structure was completely replaced by various sizes of fluid-filled cysts (arrow pointed). (C) Higher magnification power view of the cystic kidney from mouse EH85 showing that the renal tubules and glomeruli were compressed and atrophied. The glomerulus was enclosed and lost its capillary loop structure (arrow pointed). (D) Swelling renal tubule epithelial cells, hydropic degeneration, and vacuolation in the cells were observed (arrow pointed) in the kidney from G3TM mouse EJ08. (E) A kidney from the G3TM mouse EM06, showing the cyst surrounding flat epithelial cells (arrow pointed) that might be caused by fluid pressure changes resulting from cyst formation. (F) An abnormal glomerulus with poorly defined capillary loop (arrow pointed) in the kidney from G3TM mouse CS87.
The H&E of the kidney sections showed that the kidneys from wild type mice developed normal renal tubules and glomeruli (Figure 2A), while the kidneys from G3TM mice displayed a range of phenotypes associated with renal dysplasia and renal cyst formation. In the G3TM mouse EH85, the normal histological structure of the right kidney was completely replaced by fluid-filled cysts of various sizes(Figure 2B). At higher magnification, we could observe that the renal tubules and glomeruli were compressed and atrophied, and the glomerulus lost its capillary loop structure completely (Figure 2C). These data show the severe fluidfilled cyst formation and total loss of renal function in this kidney. In the kidney from G3TM mouse EJ08, cellular swelling or hydropic degeneration and vacuolation were observed (Figure 2D), suggesting the dysfunction of ion and water regulation in these renal cells. In the kidneys from G3TM mouse EM06, the cyst is surrounded by flat epithelial cells, which suggests that cellular morphological changes are caused by fluid pressure from the cyst (Figure 2E). In the kidney from G3TM mouse CS87, we found the abnormal glomerulus with poorly defined capillary loop (Figure 2F).The abnormal glomerulus with semi-enclosed capillary loop was also frequently observed, indicating the loss of glomerulus function and downregulation of blood filtering function.
Together these data suggest that kidneys from G3mTR-/-WRN-/-p53S/Smice were hypoplastic and developed PKD phenotypes.
The correlation of tumorigenesis and PKD phenotypes
As described earlier, the G3TM mice should manifest phenotypes that correlate with abnormal DNA damage response and abnormal proliferation. In our case, it manifested as increased tumorigenesis and PKD formation.To further understand the relationship between abnormal DNA damage response, tumorigenesis, and PKD phenotypes,we analyzed the frequencies and co-occurrence of cystic kidney and tumorigenesis in mice groups with different genotypes.
We did not find any tumorigenesis or PKD in those mice with WRN and telomerase double knockout, including G1DM mice (n=41), G2DM mice (n=52), and G3DM(n=63). However, we observed a few PKD or tumor incidences in G1TM and G2TM mice; this number increased dramatically in G3TM mice (Table 1, Figure 3A). The incidence increased along with telomere shortening (G1-G2-G3) and the introduction of p53S (TM vs. DM). These data strongly suggest that interplay of telomere DNA damage and p53S mutation contributed to the development of PKD.Furthermore, most PKD co-occurred with tumor phenotypes(Table 1, Figure 3A), showing that the occurrence of PKD phenotype was highly correlated with increased tumorigenesis.
Gene signatures of PKD caused by telomere dysfunction and p53S mutation
Since the genetic defect in this PKD model is very different from classical PKD models with polycystins defects, we were interested in investigating the gene signatures in MEFs(G3TM), cystic kidneys, and tumors from G3TM. We compared the gene expression profiles in MEFs from G1DM to G3TM mice using RNA-seq and ssGSEA analysis, as well as the tumors and cystic kidneys from G3TM mice.
First, we analyzed the gene signatures that were upregulated or downregulated in cystic kidneys using the Hallmark dataset. We found that the metabolism-related pathways, particularly lipid metabolism, were strikingly upregulated in cystic kidneys. These included bile acid metabolism, fatty acid metabolism and others (Figure 3B).Cell cycle-related pathways were clearly downregulated, such as mitotic spindle, G2M checkpoint, and E2F targets. (Figure 3B). These data suggest that abnormal metabolic regulation contributed greatly to PKD progress in G3TM mice.
Interestingly, the pathways such as oxidative phosphorylation, complement, and interferon alpha gamma were upregulated in both cystic kidneys and tumors (Figure 3B). These common regulated pathways suggest that the development of cystic kidney shares common mechanisms with tumorigenesis.
We then expanded the ssGSEA analysis by combining the Hallmark, C2, and C5 datasets20, and mapping the gene signatures that were gradually upregulated or downregulated in G3TM cells, tumors, and cystic kidneys (Supplementary Figure S1 and S2). The data revealed that most strikingly upregulated pathways shared by tumors and cystic kidneys included complement pathways, the immune response, lipid metabolism, and mitochondrial energy homeostasis.Interestingly, we observed that kidney function-related pathways, such as microvillus organization and water homeostasis, were upregulated in both tumors and kidneys.The data also show that organic cation transport andglucuronidation pathways were highly upregulated in cystic kidneys (Supplementary Figure S1).
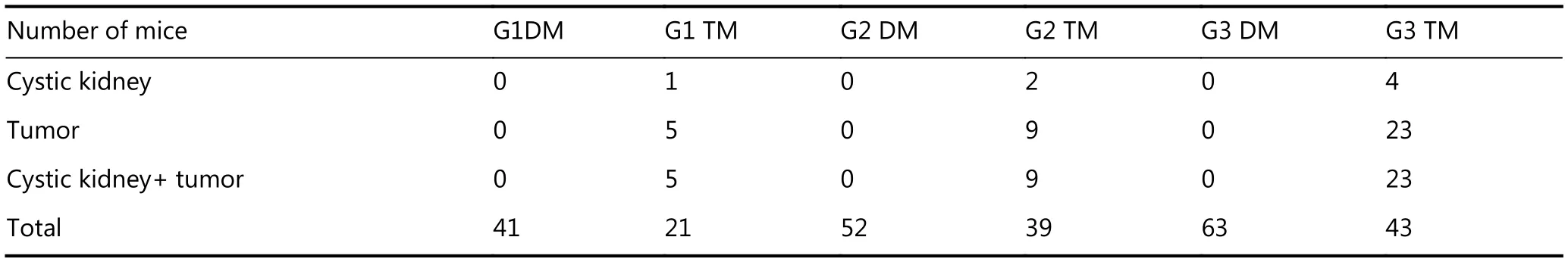
Table 1 The occurrence of cystic kidney and/or tumor in mice with different genotypes
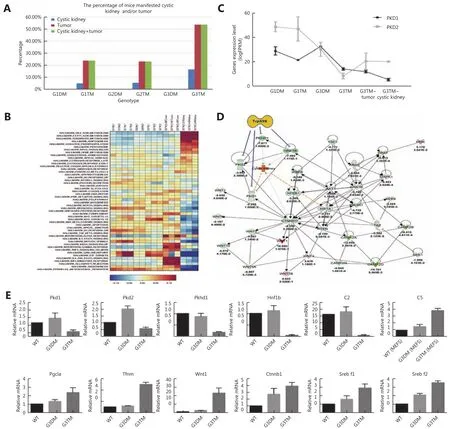
Figure 3 Co-occurrence of tumorigenesis with PKD and gene signature analysis. (A) The percentage of tumor/cystic kidney incidence in mice with genotypes from G1DM (G1mTR-/-WRN-/-) to G3TM (G3mTR-/-WRN-/-p53S/S) indicated the co-occurrence of tumorigenesis with PKD, and the incidence increased with telomere shortening (G1-G2-G3), and the introduction of p53S (TM vs. DM). (B) The heatmap of gene expression profiles (ssGSEA analysis results of the RNA-seq data using Hallmark dataset) in MEFs from G1DM, G2DM, G3DM, G1TM, G2TM,and G3TM mice, as well as the tumors and cystic kidneys from G3TM mice. The pathways were ranked by scores showing up- (red) or downregulation (blue) in cystic kidney, as well as in tumor and in G3TM MEFs. (C) The expression levels of PKD1 and PKD2 decreased significantly from G1DM to G3TM, along with the introduction of p53S mutation and telomere shortening. (D) The interaction network of genes essential for classical PKD development generated by Ingenuity Pathway Analysis (IPA). The expression fold change and P-values are shown under the gene name. Genes downregulated by Trp53S are connected by blue inhibition lines. As per IPA knowledge base, orange lines indicate gene expression level is consistent with activation of cystic kidney, whereas grey lines are inconsistent with activation of cystic kidney. (E) The validation of key gene expression levels by quantitative real-time PCR. The expression levels of genes involved in PKD pathway (PKD1, PKD2, Pkhd1, Hnf1b), complement pathway (C2 and C5), mitochondria pathway (Pgc1a and Tfam), Wnt signaling pathway(Wnt1 and Ctnnb1), and lipid metabolism pathway (Srebf1 and Srebf2) were evaluated by real-time PCR in G3TM MEFs. WT MEFs and G3DM MEFs were used as controls.
On the other hand, the pathways obviously downregulated in tumor and kidneys included cytoskeleton regulation,extracellular signal transduction and others (Supplementary Figure S2). Together, regulation of these pathways revealed that G3TM PKD shares common mechanisms with tumorigenesis. These dysfunctions of gene regulation composed the gene signatures of G3TM PKD.
Comparison of gene signatures in PKD caused by telomere dysfunction and p53S mutation with classical PKD caused by PKD1 or PKD2 deficiency
After analyzing the gene signatures in the G3TM PKD model(G3mTR-/-WRN-/-p53S/S), we compared the gene signatures in this model with classic PKD models with PKD1 or PKD2 deficiency. We analyzed RNA-seq data of the classic PKD models with PKD1 or PKD2 deficiency22by the same ssGSEA analysis, and compared gene signatures between the three mouse models. The data, analyzed by the Hallmark dataset,showed that the common upregulated pathways among these three PKD models included complement, coagulation, and apical surface, whereas the common downregulated pathways included angiogenesis (Supplementary Table S1).
The expanded analysis with Hallmark, C2, and C5 datasets revealed that common upregulated pathways included complement activation, bile acid metabolism, and ion homeostasis. The common downregulated pathways included cell-to-cell adhesion signaling and epithelial structural maintenance (Supplementary Table S2).
Together these data reveal that although the G3TM PKD model was derived from different genetic aberrations to classical PKD models, they share common pathways in regulating complement activation, lipid metabolism, cell-tocell adhesion signaling etc. These pathways might play an essential role in PKD development.
Furthermore, we found that from G1DM to G3TM, along with the introduction of p53S mutation and telomere shortening, the expression levels of PKD1 and PKD2 decreased significantly (Figure 3C), suggesting that p53S mutation could downregulate PKD1 and PKD2 expression.In the end-stage tumor and cystic kidney tissues, the PKD2 level was slightly upregulated, but was still lower than the level in G3DM (Figure 3C).
Since G3TM is the genotype with most incidences of tumor and cystic kidney disease, but not G3DM, comparison of gene regulation in G3TM with G3DM might provide the mechanisms for PKD attributed to p53S. We evaluated the genes essential for classical PKD development1,21, and mapped their interaction networks with IPA (Figure 3D).Based on expression fold-changes of genes in this interaction network, the molecule activity predictor showed that cystic kidney module was significantly activated (P-value: 3.31E-11). Other than PKD1 and PKD2, the ARPKD protein Pkhd1(polyductin) and its transcriptional factor Hnf1b (hepatocyte nuclear factor 1 homeobox B)23were also downregulated.These data suggest that p53S plays a role in transcriptional regulation of PKD-related genes.
To validate the key genes in altered pathways as revealed by RNA-seq data, we further analyzed the regulation of genes involved in the PKD pathway, complement pathway,mitochondria pathway, Wnt signaling pathway, and lipid metabolism pathway by quantitative real-time PCR.Compared with WT and G3DM MEFs, the expression of PKD genes PKD1, PKD2, Pkhd1, and Hnf1b was suppressed in G3TM MEFs. However, complement pathway genes C2 and C5; mitochondria pathway genes Pgc1a and Tfam; Wnt signaling pathway genes Wnt1 and Ctnnb1; and lipid metabolism pathway genes Srebf1 and Srebf2 were upregulated in G3TM MEFs (Figure 3E). These data further confirmed the RNA-seq data, and suggest that p53S regulates genes involved in the aforementioned pathways attributed to the development of cystic kidney.
Discussion
It has always been suspected that the development of cystic kidney disease shares features with tumorigenesis, although the evidence is unclear24,25. Recent understanding of aberrant downstream pathways in ADPKD demonstrates that transcriptional functions that regulate cell cycle progression,energy metabolism, and secretion-related signaling are abnormal in PKD1, and p53 is the essential node in all these transcriptional regulations26.
It has always been documented that wild type p53 could bind to the PKD1 promoter, and the kidneys of p53 null mice expressed higher PKD1 mRNA levels than wild-type littermates, suggesting that wild type p53 suppressed the expression of PKD14. It has also been shown that depletion of PKD1 led to increased cell proliferation and caused a premature G1/S transition, and the elevated expression of mechanosensory polycystins in human carotid atherosclerotic plaques was associated with p53 activation6,27.Thus, it is conceivable that mutant p53, which loses the wild type function of p53 and gains oncogenic function, plays an important role in the development of PKD.
Here we revealed a novel PKD and tumor combined mouse model (PKD derived from G3mTR-/-WRN-/-p53S/Smice) (Figure 1 and 2). The co-occurrence of cystic kidneys and tumors suggests common genetic mechanisms, which in this case could be DNA damage caused by telomere dysfunction and the abnormal DNA damage response,cellular proliferation, or metabolic dysregulation caused by p53N236S mutation. This model provides direct evidence to connect mutant p53 DNA damage response with PKD development. The fact that the incidences of cystic kidneys increased along with telomere shortening suggests that DNA damage triggered the development of PKD.
To dissect the common genetic causes of PKD and tumorigenesis, we identified the upregulated pathways in tumors and cystic kidneys. Among the common pathways in cystic kidneys and tumors, the pathways of activation of complement, inflammatory response, and mitochondrial function were most significantly upregulated (Figure 3B and 3E, Supplementary Figure S1). It has been documented that activation of the alternative complement pathway and the consequent inflammatory response plays an essential role in the progress of kidney diseases, such as atypical hemolytic uremic syndrome, C3 glomerulopathies, and atypical post infectious GN, as well as ADPKD28,29. These data suggest the importance of complement cascade in the regulation of inflammatory response of both cystic kidney disease and tumors.
Mitochondrial function is essential in energy metabolism,oxygen consumption, ROS regulation, and ATP synthesis.Aside from kidney disease, mitochondrial dysfunction is also related to the processes of aging and tumor development30,31.By ssGSEA analysis, we found that the pathways involved in mitochondrial function and related fatty acid metabolisms are highly activated in tumors and cystic kidneys from G3TM mice; however, they are not significantly up-regulated in PKD1- or PKD2-deficient PKD22(Supplementary Table S1,S2).
It is very promising that we found that PKD1, PKD2,Pkhd1, and Hnf1b were all downregulated by the introduction of p53S (Figure 3D). It has been documented that Hnf1b is the transcription factor for both Pkhd1 and PKD2. Mutation of Hnf1b results in kidney phenotypes that include renal agenesis, dysplasia, and cysts32. These phenotypes are consistent with our pathological analysis(Figure 2).
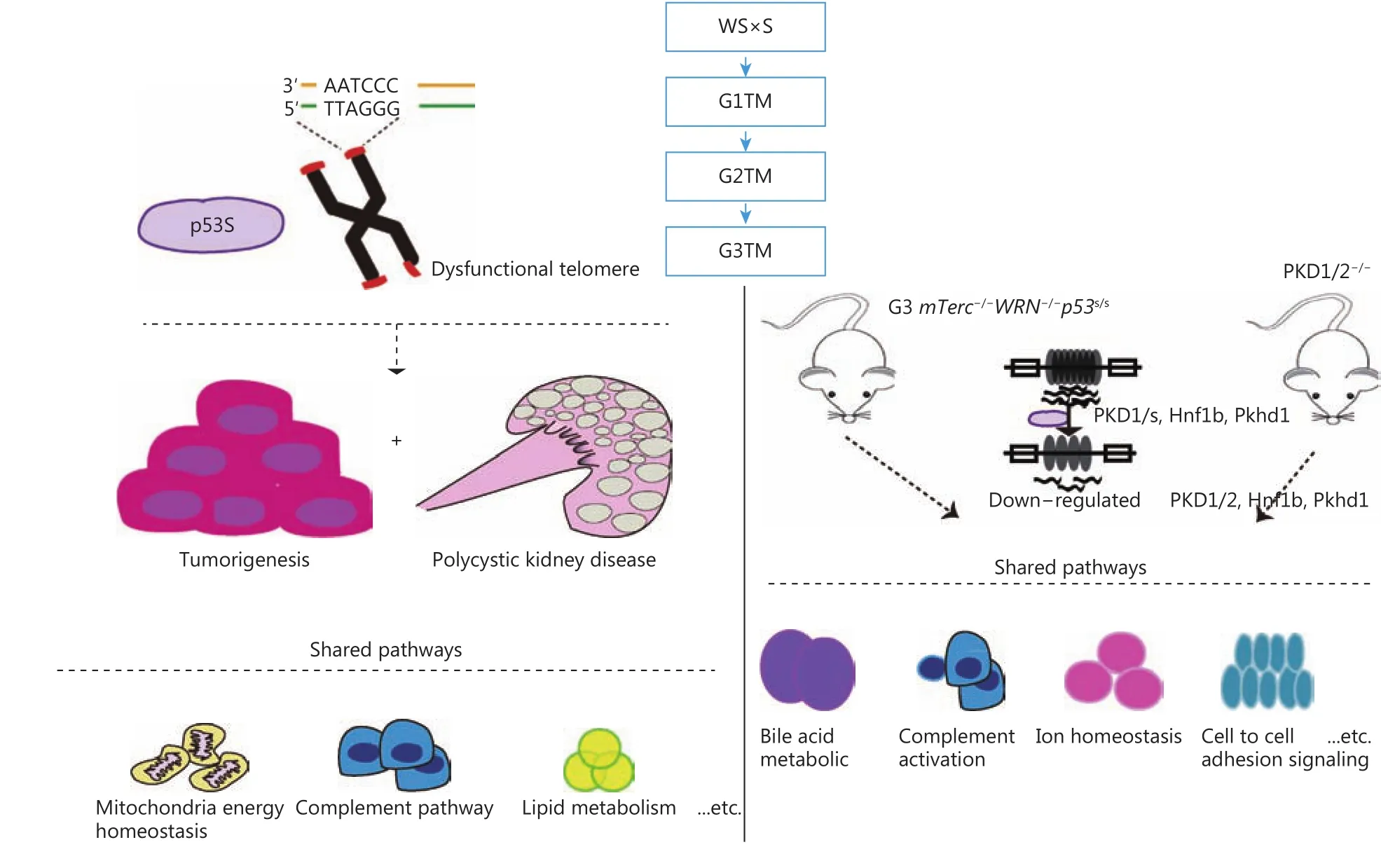
Figure 4 A schematic of the establishment of G3TM PKD model, and the gene signatures shared between development of PKD and tumorigenesis, and with PKD1/2 PKD model.
Putting these data together, we report a novel PKD and tumor combined mouse model, and reveal the gene signatures involved in the development of PKD. The G3TM PKD model shared common pathways with classical PKD.These common pathways might be essential in PKD progress,and thus could be common targets for PKD prevention, drug screening, and patient care strategies. In depth analyses of these pathways could provide new biomarkers for the clinical diagnosis and prognosis of PKD (Figure 4).
Acknowledgements
This work was supported by National Natural Science Foundation of China (NSFC) (Grant No. 30771194 and 31170735). We thank Dr. Sandy Chang from Yale University and Dr. Ronald A Depinho from The University of Texas MD Anderson Cancer Center for kindly providing WS mice.
Conflict of interest statement
No potential conflicts of interest are disclosed.
 Cancer Biology & Medicine2019年1期
Cancer Biology & Medicine2019年1期
- Cancer Biology & Medicine的其它文章
- Application of next-generation sequencing technology to precision medicine in cancer: joint consensus of the Tumor Biomarker Committee of the Chinese Society of Clinical Oncology
- The breakthrough in primary human hepatocytes in vitro expansion
- Circular RNAs and human glioma
- Qidong: a crucible for studies on liver cancer etiology and prevention
- The PI3K/Akt/GSK-3β/ROS/eIF2B pathway promotes breast cancer growth and metastasis via suppression of NK cell cytotoxicity and tumor cell susceptibility
- Estrogen and insulin synergistically promote endometrial cancer progression via crosstalk between their receptor signaling pathways