
Full-length mRNA sequencing in Saccharina japonica and identification of carbonic anhydrase genes
2019-03-22 02:29YanhuiBiJialiLiZhigangZhou
Aquaculture and Fisheries 2019年2期
Yanhui Bi,Jiali Li,Zhigang Zhou,∗
aKey Laboratory of Exploration and Utilization of Aquatic Genetic Resources,Ministry of Education,Shanghai Ocean University,Shanghai,201306,China
bInternational Research Center for Marine Biosciences at Shanghai Ocean University,Ministry of Science and Technology,Shanghai,201306,China
cNational Demonstration Center for Experimental Fisheries Science Education,Shanghai Ocean University,Shanghai,201306,China
Keywords:Carbonic anhydrase Mutigene family Single-molecule real-time sequencing Full-length mRNA sequencing Saccharina japonica
ABSTRACT The carbonic anhydrases(CAs)are a group of enzymes that play an important role in the absorption and transportation of CO2in Saccharina japonica.They are encoded by a superfamily of genes with seven subtypes that are unrelated in sequence but share conserved function in catalyzing the reversible conversion of CO2and HCO3-.Here we have characterized the CA members in the transcriptome of S.japonica using Single-molecule real-time(SMRT)sequencing technology.Approximately 9830.4 megabases from 5,028,003 quality subreads were generated,and they were assembled into 326,512 full-length non-chimeric(FLNC)reads,with an average fl nc read length of 2181 bp.After removing redundant sequences,79,010 unique transcripts were obtained of which 38,039 transcripts were successfully annotated.From the full-length transcriptome,we have identified 7 full-length cDNA sequences for CA genes(4 α-CAs,1 β-CAs and 2 γ-CAs)and assessed for their potential functions based on phylogenetic analysis.Characterizations of CAs will provide the ground for future studies to determine the involvement of CAs in inorganic carbon absorption and transportation in S.japonica.
1.Introduction
Saccharina japonica is the most economically important seaweed cultivated in Eastern Asian countries especially in China.In 2016,Saccharina cultivation covered approximately 44,000 ha with an annual production higher than one million tons in dry weight in China(Guo&Zhao,2017).Apart from its high economic value in the food,fertilizer,chemical,drug and phycocolloid industries,another important aspect for its large-scale production is the high biomass productivity.It has been reported that productivities of S.japonica and of other large brown algae range from 3300 to 11,300 g/m2per year dry wt(Gao&McKinley,1994).Cultivated S.japonica can exceed 15,000 g/m2dry wt(Brinkhuis,Levine,Schlenk,&Tobin,1987),which is 6.5 times higher than the maximum projected yield for sugarcane,the most productive cultivated land plant.This phenomenon suggests that S.japonica possesses a higher photosynthetic capacity than terrestrial C4 plants.Rubisco is the principal carboxylation enzyme in photosynthetic carbon fixation pathway.In the seawater,the concentration of rubisco's substrate,CO2,is about 12μmol/L,which is much lower than the halfsaturation constant(Ks)of rubisco for CO2in macroalgae(40-90μmol/L)(Kerby&Raven,1985).As a result,almost all macroalgae have evolved a carbonic anhydrases(CAs)depending carbon-concentrating mechanism(CCM)to increase concentration of CO2around rubisco to enhance the rate of carboxylation(Price,Badger,Woodger,&Long,2008;Reinfelder,2011).
CAs belong to a superfamily,which was classified into seven subtypes,α-CA,β-CA,γ-CA,δ-CA,ε-CA,ζ-CA and θ-CA on the basis of their amino acid sequence similarity(Moroney et al.,2011;Tachibana et al.,2011).These subtypes do not share sequence homology as well as epitopes(Lane,Saito,&George,2005),but their functions are conserved.They are all zinc-metalloenzymes that catalyze the reversible conversion of CO2and HCO3-(Badger&Price,1994).Over the last decade,a wide variety of genome sequencing studies demonstrated that multiple CA isoforms existed in plants.For example,in the Arabidopsis thaliana genome 19 genes encoding CAs,which included eight α-,six βand five γ-CAs,were identified(Fabre,Reiter,Becuwe-Linka,Genty,&Rumeau,2007).In Chlamydomonas reinhardtii,a microalga model,at least 12 genes exist(three α-,six β-,and three γ-or γ-like CAs,Moroney et al.,2011)and in Phaeodactylum tricornutum and Thalassiosira pseudonana 9( five α-,two β-and two γ-)and 13(three α-, five γ-,four δand one ζ-)CA genes were identified(Tachibanana et al.,2011).CA genes have also been identified in a number of macroalgae.For example,two α-,three β-and one γ-CAs were reported in Pyropia haitanensis(Chen,Dai,Xu,Ji,&Xie,2016),as well as single α-CA(Zhang,Yang,&Wang,2010)and single β-CA in P.yezoensis(Chen et al.,2016).In S.japonica only a unique α-CA was described(Ye et al.,2014).To better understand the CCM in S.japonica,it is necessary to characterize the CA members.
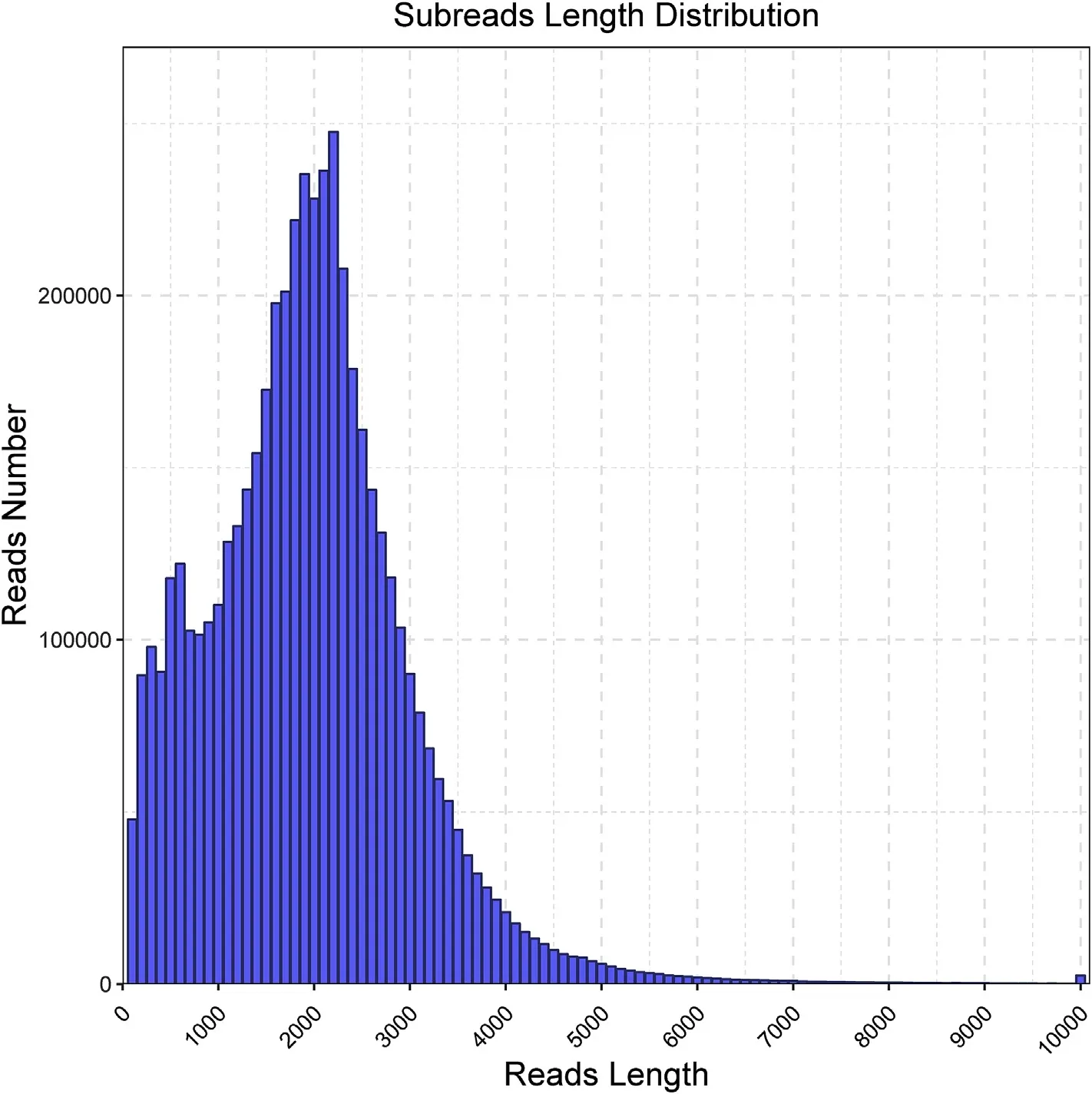
Fig.1.Length distribution of subreads.The average length of subreads is 1910 bp.
The Single Molecule Real Time(SMRT)platform was introduced by Pacific Bio-sciences for DNA sequencing(Eid et al.,2009).Compared to high-throughput sequencing platforms of Roche's 454 pyrosequencing,Illumina sequencing platform,SOLiD sequencing platform,and the Ion Torrent platform,this sequencing technology produces longer-reads of kilo-base in size,as well as avoids an assembly process and inclusion of possible errors that may be derived from shorter-reads(Westbrook,Karl,Wiseman,&Mate,2015).The processes of generating circular consensus sequence(CCS),an iterative clustering for error correction(ICE),and quality filtering(Quiver)provided by Pacific Biosciences generate high quality,full-length polished isoforms(Chin et al.,2013).In this study,we used PacBioSMRT sequencing technology to analysis the full-length transcriptome of S.japonica.In combination with the publicly available next generation sequencing(NGS)-transcriptome(Ye et al.,2015),CA subfamily gene diversity of S.japonica was studied by clustering algorithms and phylogenetic analyses.Isolation of full-length transcripts will also be useful for future genome annotation and functional studies in this alga.
2.Materials and methods
2.1.Gametophytes preparation
Gametophytes of S.japonica of the Rongfu strain were used.The female and male gametophytes were kept in the lab and cultivated in PES medium(Starr&Zeikus,1993)under vegetative growth conditions of 30 μmol photos/m2·s at(17 ± 1) °C with a photoperiod of 12:12 light/dark(Zhou&Wu,1998).The culture medium was replaced every 2 weeks.
2.2.RNA extraction
Total RNAs from female and male gametophytes were extracted using the TRIzol reagent(Invitrogen,USA).RNA integrity number(RIN)was assessed using the Agilent RNA 6000 Nano Kit(Agilent Technologies)and Agilent 2100 Bio analyzer(Agilent Technologies).The high-quality total RNA was used for library construction and sequencing.
2.3.PacBio library preparation and sequencing
Poly(A)mRNAs were isolated from total RNAs using oligo-dT magnetic beads(Dynal).cDNAs were synthesized using the SMARTer®PCR cDNA Synthesis Kit(Clontech,USA).The cDNAs were partitioned into five size-ranges of<0.5,0.5-1,1-2,2-3,and>3kb using the BluePippinSize Selection System protocol as described by Pacific Biosciences(PN 100-092-800-03).Subsequently,both ends of the cDNAs were ligated with the SMARTer®adaptors and SMRTbell to produce circular type single DNA molecules,which were sequenced using the P4-C2 chemistry of PacBio®RSII system(DNA Link,Korea).
2.4.Data processing
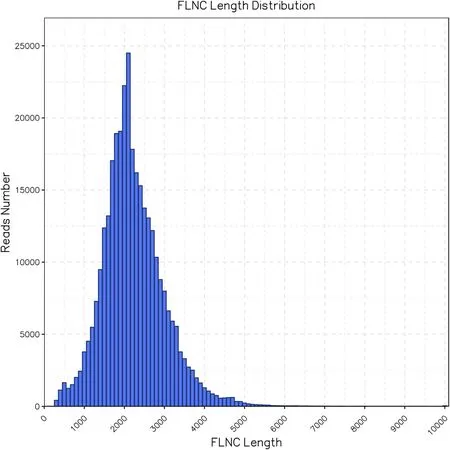
Fig.2.Length distribution of FLNC reads.The average length of fl nc reads is 2181 bp.
Subread BAM fi les were generated by processing sequence data using the SMRTlink 5.0 software.CCS was created from subread BAM fi les by searching for the presence of sequencing adapters.According to whether there are 5′and 3′cDNA primers,as well as the presence of a polyA tail preceding the 3'primer,the resulted CCSs were classified in full length and incomplete reads.To identify transcript clusters the ICE that performs a pairwise alignment and reiterative assignment of fulllength reads to clusters based on likelihood was used.After ICE,the incomplete reads were added to the isoform clusters to increase sequence coverage to obtain a final consensus using the Quiver algorithm(Chin et al.,2013).The CD-HIT program was used to reduce sequence redundancy with a parameter of 99%.
2.5.Gene functional annotation
Using BLASTX homologies searches,unigenes were annotated by comparison with the sequences deposited in non-redundant protein(nr)NCBIdatabasewithacut-off lowerthanE-value<10-5.The InterProScan program(Zdobnov&Apweiler,2001)was employed to analysis functional classi fication of Gene Orthology(GO).Classi fication of Orthologous Groups(COG)was performed using BLASTX program(E-value<10-5)(Tatusov et al.,2003)against the COG database.Pathway analysis was conducted using Kyoto Encyclopedia of Genes and Genomes(KEGG)annotation service KAAS(Kanehisa,Goto,Kawashima,Okuno,&Hattori,2004;Moriya,Itoh,Okuda,Yoshizawa,&Kanehisa,2007).
2.6.Sequences and phylogenetic analyses of CA genes
The CA genes of S.japonica were screened from the transcriptome data.Their coding nucleotide sequences(CDS)and corresponding amino acids were predicted using ORF Finder https://www.ncbi.nlm.nih.gov/orffinder/.Based on the predicted amino acid sequences their subcellular localizations were analyzed using TargetP 1.1 server http://www.cbs.dtu.dk/services/TargetP/.Phylogenetic analysis was conducted using the S.japonica CA nucleotide sequences with their homologues from diverse organisms retrieved from the NCBI database.Multiple sequence alignments were performed in MEGA 6.06 software(Tamura,Stecher,Peterson,Filipski,&Kumar,2013).The Kimura twoparameter model(Kimura,1980)was selected to compute the evolutionary distances.The Neighbor-joining(NJ)tree was generated using MEGA 6.06(Tamura et al.,2013)with 1000 bootstrap replicates.
3.Results
3.1.Transcriptome
We have obtained 5,028,003 subreads from S.japonica gametophytes with an average subreads length of 1910 bp and overall length of 9830.4 megabases(Fig.1).These subreads formed 433,480 CCSs after merging and error correction.Of the 433,480 CCSs,326,512 were found to be FLNC reads with an average length of 2181 bp(Fig.2).ICE tool was used for a further round of error correction,and 180,069 transcripts were obtained.To reduce sequence redundancy,we used CD-HIT software to cluster the consensus reads and 79,010 unigenes were obtained.
3.2.Unigenes annotation
All sequences were annotated based on nr,GO,KEGG,and KOG databases using the BLASTx algorithm and 38,039(48.1%),22,537(28.5%),29,437(37.3%),and 17,584(22.2%)genes showed significant sequence matches respectively.The majority of unigenes(21,814,57.35%)were similar to Ectocarpus siliculosus,followed by Coccomyxa subellipsoidea(869,2.28%),S.japonica(836,2.20%),Gossypium hirsutum(803,2.11%)and amongst others(13,717,36.06%)(Fig.3).
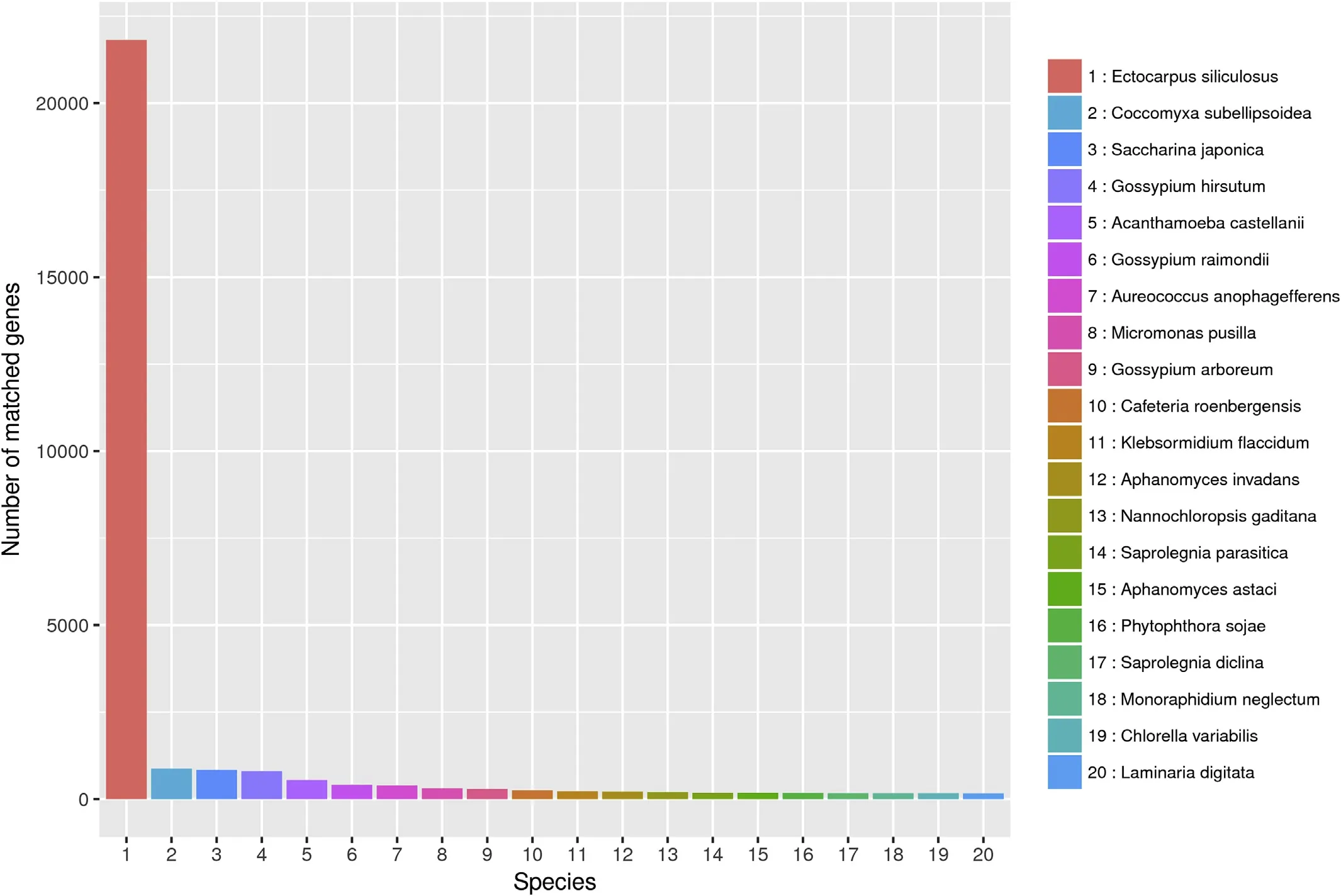
Fig.3.Species distribution of unigenes based on the BLASTX results against the NCBI nr protein database.
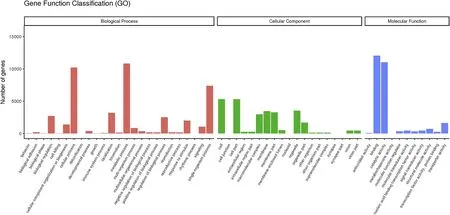
Fig.4.Gene function classi fication based on GO analysis.22,537 unigenes were assigned to 55 functional groups and classified in three categories.
Searching the GO database,22,537 unigenes were assigned to 55 functional groups within three categories.25,19,11 GO functional groups were assigned in “biological process”,“cellular component”,and“molecular function”categories,respectively.The “biological process”,“metabolic process”(48.1%),and “cellular process”(45.3%)were the most represented.In the “cellular component”category,the subcategories “cell”(23.6%)and “cell part”(23.6%)were the most represented.Within the “molecular function”category,most of the sequences were classified in the subcategories “binding”(53.4%)and“catalytic activity”(48.9%)(Fig.4).
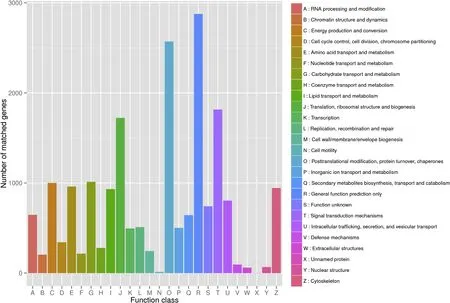
Fig.5.COG classi fication.17,584 unigenes were assigned to 26 functional categories.
Searching the COG database,17,584 unigenes were assigned to 26 functional categories,and most of the sequences were classified within the subcategories“posttranslational modi fication,protein turnover,chaperones”,“signal transduction mechanisms”and “translation,ribosomal structure and biogenesis”(Fig.5).
A total of 29,437 unigenes were assigned to 377 KEGG pathways.The majority of unigenes were assigned to“Metabolism”category of“Energy metabolism”(1677),“Carbohydrate metabolism”(1600),the“Genetic information processing”category of “Translation”(1619),“folding,sorting and degradation”(1181),and the “Environmental Information Processing”category of “Signal transduction”(1578)(Fig.6).
3.3.CA family members in S.japonica
Seven putative CAs were identified from the S.japonica SMRT-transcriptome:3 CAs(2α-and 1 γ-CA)were identical to transcripts retrieved from the NGS-transcriptome,and 4(2α-,1β-and 1 γ-CA)were newly identified(Table 1).Combination of these two transcriptome datasets,identified a total of 11 CAs in S.japonica.Except for SjβCA2,the full-length CDSs for all the CAs were found.SjαCA2,SjαCA3,SjαCA5 and two SjβCAs were secreted proteins and were probably localized in the periplasmic space,cell wall or cytomembrane.Sjα-CA1was previously described in chloroplast(Ye et al.,2014)and SjαCA4 was also predicted to be located in this organelle.All SjγCAs were predicted to be located in the mitochondrion(Table 1).
3.4.Phylogenetic analysis of CA genes
Phylogenetic analysis revealed that CAs of S.japonica and the sequence homologues clustered in three independent branches according to their subtypes with high bootstrap support.In the α-CA branch,four S.japonica α-CAs clustered with the α-CA from L.digitata and SjαCA3 clustered with Mialpha CA2 of Lobosphaera incise.In the β-CA branch,all three S.japonica β-CAs clustered with β-CAs from diatom and green alga,and then with β-CAs from red alga.All γ-CAs clustered in a single branch(Fig.7).
4.Discussion
4.1.Characterization of protein-coding genes in S.japonica transcriptomes
Isolation of full-length mRNA transcripts is crucial to understand gene function.The genome of S.japonica is approximately 537-Mb in size and encodes for 18,733 protein-coding genes(Ye et al.,2015).Other reported S.japonica transcriptome dataset predicted 19,040 unigenes(Liang et al.,2014).In this study the double of protein-coding genes were annotated and our transcriptome data provides a valuable contribution to understand the genome organization and genes of S.japonica for future studies.GO analyses revealed that in both transcriptomes,most of the transcripts were annotated in the functional category “biological process category”and within the subcategory
“binding”.Similarly,COG analysis,classified the majority of the transcripts within the“posttranslational modi fication,protein turnover,chaperones”category for both transcriptomes.KEGG analysis showed that the largest gene number was within “Translation”pathway of“Genetic information processing”.
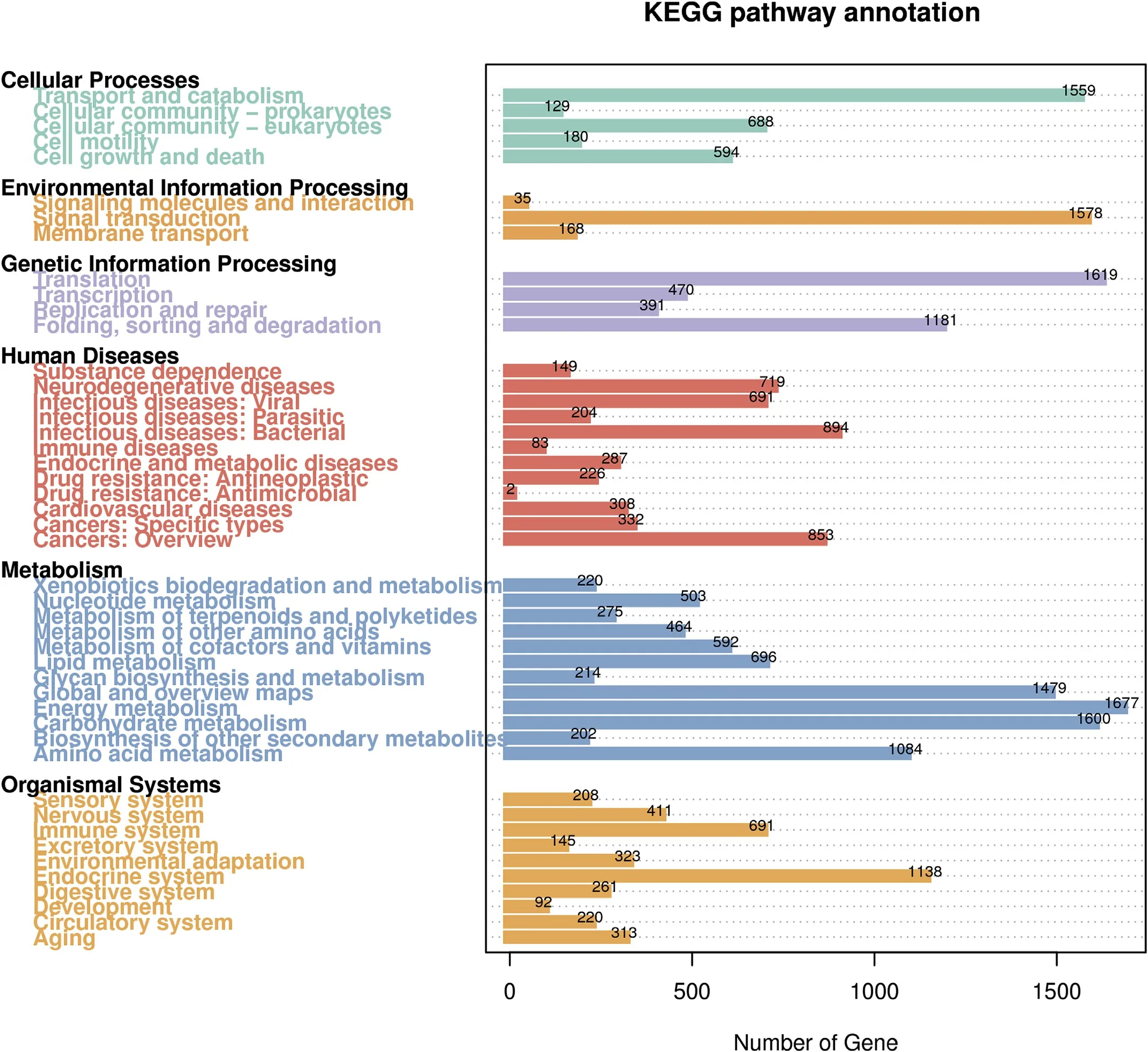
Fig.6.Metabolic pathway analysis using KEGG.29,437 unigenes were assigned to 377 KEGG pathways.
4.2.CAs of S.japonica
A total of 11 CAs were identified from S.japonica,of which 4 were newly identified transcripts.However,four CAs previously identified in the NGS-transcriptome were not found in our dataset which might be due to the different sequencing techniques.In SMRT,low abundance genes are often missed,while in NGS,genes with complex structures such as G/C-rich,G/C cluster,or poly A/T/C/G generally are not successfully sequenced.Thus,combination of NGS-and SMRT-transcriptome data might be necessary to obtain full information about multigene families.In the previous report,12 CAs(7 α-CAs,2β-CAs,and 3 γ-CAs)were screened from NGS-transcriptome of S.japonica(Bi&Zhou,2016),but only 3 α-CAs,2β-CAs,and 2 γ-CAs were veri fi ed by PCR.
Till now,subcellular locations of CAs have been intensively studied in the model organism C.reinhardtii.Among the reported 12 CA isoforms in C.reinhardtii,two α-CAs(CAH1 and CAH2)and two β-CAs(CAH7 and CAH8)were located in periplasmic space;two β-CAs(CAH4 and CAH5)and all three γ-or γ-like CAs were located in mitochondrion;one α-CA(CAH3)was located in thylakoid lumen;one β-CA(CAH6)was located in chloroplast stroma,and another one β-CA(CAH9)was located in cytoplasm(Moroney et al.,2011).Phylogenetic analysis revealed that SjαCA3 clustered with CAH1 and CAH2 of C.reinhardtii suggesting that it might also be located in periplasmic space and subcellular location prediction showed that it was a secreted protein.Similarly,in agreement with the bioinformatics predictions SjβCA1 and SjβCA2 were secreted proteins and clustered with CAH7 and CAH8 of C.reinhardtii.Moreover, γ-or γ-like CAs of C.reinhardtii and all three SjγCAs were predicted in the mitochondria.Considering the functions of CAs will depend on their subcellular locations,the related predictions above will be in the future confirmed using immunocolloid gold electron microscopy as described in Ye et al.(2014).These results combined with CA enzyme activity and transcript profile characterization will help to understand the mechanism of inorganic carbon absorption and transportation in S.japonica.
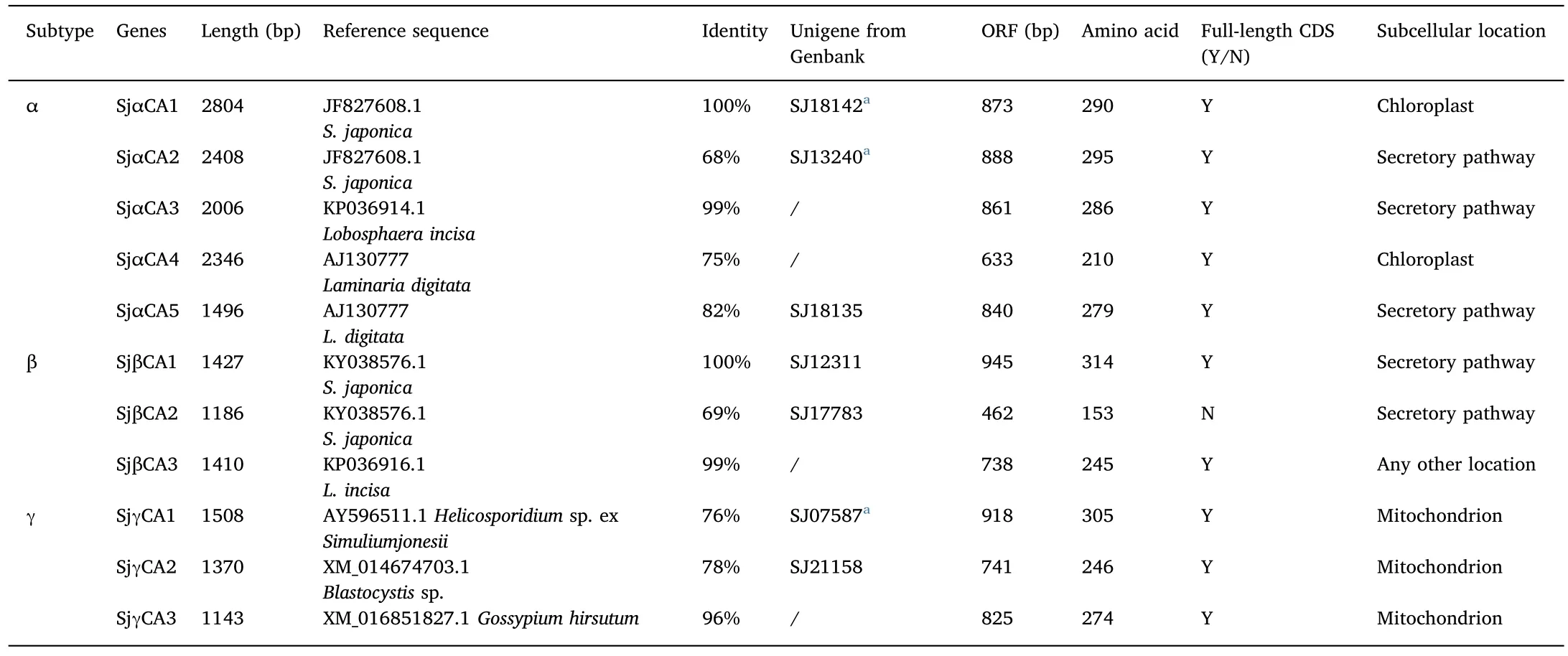
Table 1Characterizations of CA gene family obtained from SMRT-transcriptome and NGS-transcriptome of S.japonica.
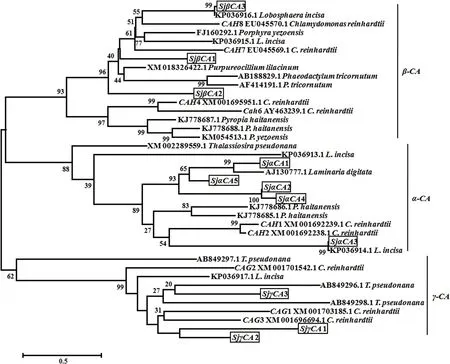
Fig.7.Phylogenetic analysis of S.japonica CA genes using Neighbor-joining(NJ)method.Bootstrap values(1000 replicates)are shown at the nodes.
Acknowledgements
Thisresearch wassupported byShanghaiUniversitiesPeak Discipline Project of Aquaculture and “Double First-Class Initiative”program for its First-Class Discipline of aquaculture.The authors declare no conflicts of interest.The authors are responsible for the contents of this paper.
 Aquaculture and Fisheries2019年2期
Aquaculture and Fisheries2019年2期
- Aquaculture and Fisheries的其它文章
- Estimationof genetic parameters for juvenile growth performance traits in olive flounder(Paralichthys olivaceus)
- Effects of dietary lysolecithin(LPC)on growth,apparent digestibility of nutrient and lipid metabolism in juvenile turbot Scophthalmus maximus L.
- In vivo effects of neomycin sulfate on non-specific immunity,oxidative damage and replication of cyprinid herpesvirus 2 in cruciancarp(Carassius auratus gibelio)
- Effectiveness of stocking Sparus macrocephalus fry in situ in the East China Sea
- Current status of research on aquaculture genetics and genomicsinformation from ISGA 2018