
Choroid plexus tumor necrosis factor receptor 1:a new neuroin flammatory piece of the complex Alzheimer's disease puzzle
2019-03-15 07:45SophieSteelandRoosmarijnVandenbroucke
中国神经再生研究(英文版) 2019年7期
Sophie Steeland , Roosmarijn E. Vandenbroucke ,
1 VIB Center for In flammation Research, Ghent, Belgium
2 Department of Biomedical Molecular Biology, Ghent University, Ghent, Belgium
Abstract Due to the aging of the population and despite the enormous scientific effort, Alzheimer's disease remains one of the biggest medical and pharmaceutical challenges in current medicine. Novel insights highlight the importance of neuroin flammation as an undeniable player in the onset and progression of Alzheimer's disease. Tumor necrosis factor is a master in flammatory cytokine that signals via tumor necrosis factor receptor 1 and tumor necrosis factor receptor 2, but that also regulates several brain functions in health and disease. However, clinical trials investigating drugs that interfere with the tumor necrosis factor pathway in Alzheimer's disease led to inconclusive results, partially because not only the pro-in flammatory tumor necrosis factor/tumor necrosis factor receptor 1, but also the beneficial tumor necrosis factor/tumor necrosis factor receptor 2 signaling was antagonized in these trials. We recently found that tumor necrosis factor is the main upregulated cytokine in the choroid plexus of Alzheimer's disease patients, signaling via tumor necrosis factor receptor 1. In agreement with this, choroidal tumor necrosis factor/tumor necrosis factor receptor 1 signaling was also upregulated in different Alzheimer's disease mouse models. Interestingly, both genetic and nanobody-based pharmacological blockage of tumor necrosis factor receptor 1 signaling was accompanied by favorable effects on Alzheimer's disease-associated inflammation, choroidal morphology and cognitive functioning. Here, we brie fly summarize the detrimental effects that can be mediated by tumor necrosis factor/tumor necrosis factor receptor 1 signaling in (early) Alzheimer's disease, and the consequences this might have on the disease progression. As the main hypothesis in Alzheimer's disease clinical trials is still based on the amyloid beta-cascade, the importance of Alzheimer's disease-associated neuroinflammation urge the development of novel therapeutic strategies that might be effective in the early stages of Alzheimer's disease and prevent the irreversible neurodegeneration and resulting memory decline.
Key Words: tumor necrosis factor; neuroin flammation; blood-cerebrospinal fluid barrier; preclinical research;drug development; neurodegeneration; cognitive decline; mouse models; TNFR
Alzheimer's Disease Is One of the Largest Unmet Medical Needs With the Highest Clinical Trial Failure Rate Among AllTherapeutic Areas
The devastating neurodegenerative disorder Alzheimer's disease is the leading cause of dementia in patients older than 65 years. Due to the aging of the population, it is expected that the number of Alzheimer's disease patients will triple by 2050. Moreover, despite the scientific efforts, there is currently no preventive, delaying or curative therapy.Neuroin flammation is a typical hallmark of Alzheimer's disease and is now recognized as being a key player that drives the onset and progression of the disease (Ransohoff, 2016).Tumor necrosis factor is one of the best studied pro-in flammatory cytokines in systemic, in flammatory disorders, however, its pleiotropic actions in the brain have only recently begun to be acknowledged (Steeland et al., 2018a). It was found that tumor necrosis factor, a cytokine that signals via tumor necrosis factor receptor (TNFR)-1 and TNFR2, regulates brain function in health and disease. Generally, TNFR1 is considered to be the mediator of tumor necrosis factor's pro-in flammatory and apoptotic activities, whereas signaling via TNFR2 controls regenerative and homeostatic processes (Steeland et al., 2018a). Also, TNFR1 is constitutively expressed by most cells, including brain cells in for example the hippocampus, while the expression of TNFR2 is more restricted to primarily immune cells, including microglia,endothelial cells, and some neuronal populations (Probert,2015). In Alzheimer's disease, a central role has been attributed to tumor necrosis factor due to its importance in synaptic (dys)functioning and memory formation, its co-localization with amyloid beta plaques in Alzheimer's disease brains and its contribution to amyloidogenesis (Steeland et al., 2018a). Epidemiologic studies linked the use of anti-inflammatory medication to a lower incidence of Alzheimer's disease but unfortunately, strategies targeting these in flammatory pathways have not been successful until so far. Also attempts to inhibit tumor necrosis factor in clinical trials led to inconclusive results (Tobinick, 2009). A novel approach that has been put forward only focuses on the pro-in flammatory arm of tumor necrosis factor by selectively antagonizing TNFR1 (Steeland et al., 2018a). Our own work adds to that as we identified tumor necrosis factor/TNFR1 signaling as the main upstream activated cytokine in the choroid plexus of Alzheimer's disease patients (Steeland et al., 2018b). Importantly, we confirmed this detrimental role in two Alzheimer's disease mouse models, and therapeutic abrogation of this pathway impedes the memory decline associated with this disorder. Our study further confirms the harmful effects of in flammation in Alzheimer's disease and encourages the development of new drugs directed against the early inflammatory stage of the disease. We have performed a PubMed literature search of articles published in the period of 2009 — September 2018 on neuroin flammation and the involvement of tumor necrosis factor/TNFR1 in Alzheimer's disease.
Observations in Human and Mouse Alzheimer's Disease Highlight the Importance of Choroidal Tumor Necrosis Factor Receptor 1
In our study, we stress the importance of the choroid plexus, which is an often neglected brain structure. The choroid plexus contains the blood-cerebrospinal fluid barrier consisting of choroid plexus epithelial cells. Apart from secreting cerebrospinal fluid, the choroid plexus is also involved in cerebrospinal fluid dynamics and growth factor secretion.This epithelial monolayer that is uniquely positioned at the interface between the periphery and the brain, is believed to operate as an important sensor of peripheral in flammation,and forms a selective gateway to the brain for circulating immune cells (Demeestere et al., 2015; Balusu et al., 2016). As the importance of the choroid plexus in Alzheimer's disease has already been highlighted in numerous publications (reviewed by Balusu et al. (2016)), we performed an ingenuity pathway analysis on choroid plexus tissue of late-stage Alzheimer's disease patients. This analysis revealed that tumor necrosis factor is the most upstream activated cytokine in Alzheimer's disease patients compared to healthy subjects and that a long list of NF-κB-dependent genes downstream tumor necrosis factor/TNFR1 signaling were activated (Steeland et al., 2018b). Based on these results, we focused our subsequent analyses on choroid plexus-mediated neuroinflammation while other Alzheimer's disease processes were not studied. To investigate the importance of tumor necrosis factor/TNFR1 signaling at the choroid plexus in Alzheimer's disease, we used two different amyloid beta-driven Alzheimer's disease mouse models: the acute model of intracerebroventricular injection of amyloid beta oligomers and the transgenic amyloid precursor protein (APP)/presenilin-1(PS1)tg/wtmouse model (Radde et al., 2006; Brkic et al., 2015).In both models, several pro-inflammatory genes were significantly induced in the choroid plexus and hippocampus,although the effects were more pronounced upon amyloid beta oligomers injection in wild type (WT) mice compared to late-stage APP/PS1tg/wtmice. Crossing these mouse models into a TNFR1-deficient background resulted in a clear reduction of some prominent inflammatory mediators in the choroid plexus and hippocampus (Steeland et al., 2018b)(Figure 1A-D). To link the importance of tumor necrosis factor/TNFR1 signaling in Alzheimer's disease-associated neuroinflammation with the choroid plexus, we examined the latter more closely in both Alzheimer's disease mouse models. It was previously reported that the typical morphology and functions of the choroid plexus are disturbed in Alzheimer's disease patients (Balusu et al., 2016). Indeed,Chalbot et al. already suggested in 2011 that choroid plexus epithelial damage is one of the first signs of Alzheimer's disease (Chalbot et al., 2011) and former research of our group pointed towards the destructive effects of amyloid beta oligomers on the choroid plexus epithelial architecture (Brkic et al., 2015). Consequently, we investigated this using transmission and scanning electron microscopy and confirmed the loss of the typical cuboidal structure of choroid plexus epithelial cells upon intracerebroventricular injection of amyloid beta oligomers. Intriguingly, we found that the choroid plexus epithelial morphology in TNFR1-/-mice was completely preserved. Choroid plexus epithelial cells of 18-week-old APP/PS1tg/wtmice displayed a profound transformation to a more degenerative state with irregular nuclei and signs of cellular degradation, whereas these of non-transgenic age-matched littermates exhibit a completely normal morphology. In a TNFR1-deficient background,these signs of cellular degradation were less obvious and the cuboidal shape of the cells was maintained (Figure 2).
Genetic Abrogation of Tumor Necrosis Factor Receptor 1 Prevents Amyloidogenesis and Reduces Neuroin flammation
We previously highlighted the importance of the blood-cerebrospinal fluid barrier integrity in Alzheimer's disease.In the same study, we reported that the pro-inflammatory matrix metalloproteinase-3 was responsible for blood-cerebrospinal fluid barrier breakdown (Brkic et al., 2015). In our most recent study, we found that the permeability of the blood-cerebrospinal fluid barrier was maintained in amyloid beta oligomers-injected TNFR1-/-mice thanks to lower choroidal expression of matrix metalloproteinase-3 and -8 (Figure 1E and F) (Steeland et al., 2018b). Matrix metalloproteinases are known to cleave tight junction proteins that are essential to keep the barrier integrity intact. Because changes in the choroid plexus morphology and loss of barrier integrity might favor leukocyte extravasation into the brain parenchyma, culminating in more central nervous system in flammation (Brkic et al., 2015), it has been suggested to consider matrix metalloproteinase-3 as a potential druggable target to protect barrier integrity (Brkic et al., 2015). However, despite the great efforts made by pharmaceutical companies, most developed matrix metalloproteinase inhibitors lack specificity and are broad-spectrum antagonists with unwanted side effects after prolonged treatments. Additionally, the broad spectrum of biological activities of matrix metalloproteinases hampers their inhibition: in some conditions, they can be harmful whereas they are antitargets in other conditions(Vandenbroucke and Libert, 2014). Our approach, namely targeting tumor necrosis factor/TNFR1 signaling upstream of matrix metalloproteinase activation, has the benefit that it is very selective. Moreover, our study revealed that inhibiting TNFR1 has broader therapeutic effects in Alzheimer's disease compared to matrix metalloproteinase-3 inhibition.Indeed, blocking this pathway not only prevents the Alzheimer's disease-associated inflammation and loss of barrier integrity but it also reduces the amyloidogenesis. To address this, we investigated Alzheimer's disease pathology in APP/PS1tg/wtmice in a WT and TNFR1-deficient background. This transgenic mouse model displays several typical pathological features of Alzheimer's disease such as the presence of amyloid plaques and microglia activation in the brain (Radde et al., 2006; Steeland et al., 2018b). Strikingly, we observed reduced amyloid beta pathology in APP/PS1tg/wtTNFR1-/-mice re flected by less amyloid plaques in the brain and lower levels of soluble Aβ42in the cortex compared to transgenic mice in a WT background (Figure 1G). One explanation for the latter is a reduced expression of the enzyme responsible for the generation of toxic amyloid peptides, beta-secretase(BACE1), that we observed in the transgenic TNFR1-/-mice.In line with the above observations in transgenic TNFR1-/-mice, there was also less microglia activation in their brains compared to transgenic WT mice, indicative for less in flammation and consequent neuronal cell loss (Figure 1H). Importantly, a recent study recognized a positive relationship between microglia activation and amyloid deposition which was stronger in individuals with mild cognitive impairments(Dani et al., 2018). Altogether, this argues for the importance of neuroinflammation in early phases of the disease and supports our approach that tackles these early, in flammatory events to treat Alzheimer's disease.
Genetic and Pharmacological Ablation of Tumor Necrosis Factor Receptor 1 Prevents Against Alzheimer's Disease-Induced Memory Decline
Cognitive impairment is one of the most prominent symptoms of Alzheimer's disease and is also reported in APP/PS1tg/wtmice (Radde et al., 2006). In our study, we evaluated cognitive behavior by assessing the working memory with the novel object recognition test. Both short-term and longterm memory declined in APP/PS1tg/wtmice compared to non-transgenic age-matched controls, and in WT mice upon intracerebroventricular injection of amyloid beta oligomers.Strikingly, TNFR1 deficiency improved the novel object preference and thus the working memory in both mouse models. Because all these results are promising and indicate that TNFR1 might be a potential therapeutic target, we provided proof-of-concept for therapeutic TNFR1 targeting(Steeland et al., 2018b). For this experiment, we used an inhouse generated and validated trivalent Nanobody called TROS that is able to selectively bind and block TNFR1 signaling. Co-administration of amyloid beta oligomers with this inhibitor led to encouraging results. Indeed, TROS partially prevented the amyloid beta oligomers-induced reduction in both short-term and long-term memory. These data support the idea that TNFR1 is a valuable new drug target to prevent Alzheimer's disease-associated memory decline (Figure 2).
The Beta Amyloid Hypothesis into Question
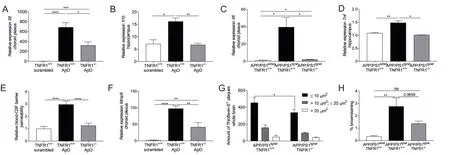
Figure 1 Neuroin flammatory effects of tumor necrosis factor receptor 1 (TNFR1) in Alzheimer's disease pathology in two Alzheimer's disease mouse models.

Figure 2 Schematic overview of the effects of choroid plexus TNFR1 in the pathology of Alzheimer's disease.
Alzheimer's disease is a complex multifactorial disorder and many hypotheses about the main cause of Alzheimer's disease exist, including the amyloid cascade, the Tau and the in flammatory hypothesis. The last few years, large advanced Phase 3 trials in Alzheimer's disease with drugs that were developed guided by the amyloid hypothesis consistently failed. Examples are the clinical trials with solanezumab developed by Eli Lilly and targeting soluble amyloid beta,and with verubecestat (Merck) that inhibits BACE1. Supporters of this hypothesis argue that Alzheimer's disease patients need to be treated earlier in the disease course but there is a growing consensus to rethink this popular amyloid hypothesis (Ransohoff, 2016). Although we used mouse models driven by amyloid beta and identified TNFR1 as a downstream effect of amyloid beta, we mainly focused on the effects that toxic soluble amyloid beta oligomers species exert early in the pathogenesis of Alzheimer's disease(Figure 2). In the acute model, we inject soluble amyloid beta oligomers directly into the ventricles, representing the early phases of Alzheimer's disease in which no plaques are formed yet. Nevertheless, also the other Alzheimer's disease hypotheses should be taken into account. Indeed, compelling research indicates that in flammation is the initiator of Alzheimer's disease instead of a simple consequence of the damage caused by accumulated amyloid beta plaques (Ransohoff, 2016). Another argument that favors this hypothesis is the contribution of lifestyle factors such as diet and exercise to Alzheimer's disease. Obesity and diabetes are associated with a higher inflammatory status, which promotes the formation of amyloid plaques (Sohn, 2018). Altogether,this indicates that prodromal Alzheimer's disease patients might benefit from therapeutics that target the inflammatory component of the disease. Cutting the inflammation mediated by tumor necrosis factor/TNFR1 has the advantage that it will interfere with the complex amyloidogenesis process, in addition to its direct anti-inflammatory effects.Furthermore, the normal choroid plexus morphology will be maintained and blood-cerebrospinal fluid barrier integrity ensured, limiting the contribution of the peripheral immune compartment. Thus early blockage of this pathway might possibly overcome the neurodegenerative progression and prevent memory deterioration. In future research, efforts should be put to gain insights in the exact role of the choroid plexus in this complex pathology, and whether selective blockage of choroidal TNFR1 is sufficient to overcome the pathological features in Alzheimer's disease. This would be interesting from a therapeutic perspective, as the choroid plexus is easily accessible for peripheral drugs via its fenestrated stromal capillaries, and thus could form the basis for new anti-in flammatory Alzheimer's disease drugs.
Author contributions:Manuscript drafting: SS, manuscript editing and final version permitting: REV.
Conflicts of interest:None declared.
Financial support:This work was supported by the Research Foundation Flanders (FWO), The Foundation for Alzheimer's Research Belgium (SAOFRA) and European Union Cost action MouseAge (BM1402), and the Baillet Latour Fund (all to SS and REV).
Copyright license agreement:The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Hans-Gert Bernstein, Otto-von-Guericke University Magdeburg, Germany.
Additional file:Open peer review report 1.

- 中国神经再生研究(英文版)的其它文章
- Effects of Ginkgo biloba extract EGb761 on neural differentiation of stem cells offer new hope for neurological disease treatment
- Redistribution of nerve strain enables end-to-end repair under tension without inhibiting nerve regeneration
- Transcriptional dysregulation in neurodegenerative diseases: who tipped the balance of Yin Yang 1 in the brain?
- Magnesium: pathophysiological mechanisms and potential therapeutic roles in intracerebral hemorrhage
- Bridging larger gaps in peripheral nerves using neural prosthetics and physical therapeutic agents
- Exogenous neural stem cell transplantation for cerebral ischemia