
PI3K/AKT信号通路在促进口腔鳞癌侵袭转移中的作用
2019-03-12 08:12赵晓苇周静萍毕于蓝汪佳颖王维康李先振
皖南医学院学报 2019年1期
赵晓苇,周静萍,毕于蓝,汪佳颖,于 瑞,王维康,李先振
(皖南医学院 口腔医学院,安徽 芜湖 241002)
口腔鳞癌占头颈部恶性肿瘤发病率第1位[1],其恶性程度较高,浸润性较强,易于复发和转移是其致死的主要原因[2]。晚期口腔鳞癌多呈浸润性生长甚至远处转移,外科手术难以完全切除,而且传统的化疗效果也难以满意。上皮间质转化(epithelial-mesenchymal transition,EMT)已被证明在促进原发肿瘤的侵袭和转移中起着关键的作用[3],前期研究结果显示:TNF-α可以通过稳定表达转录因子snail促进口腔鳞癌细胞EMT的发生[4],但其过程中有众多信号转导通路的参与。
异常激活的PI3K/AKT信号通路已被证实与多种肿瘤的发生发展密切相关。PI3K激活的关键因子AKT可以通过磷酸化作用于其下游靶蛋白Bad、caspase9、mTOR等从而促进细胞增殖,因此AKT在抗细胞凋亡中起到重要作用[5]。
多项研究表明PI3K/AKT信号通路均可上调核转录因子snail的表达诱导肿瘤细胞发生EMT,促进肿瘤细胞发生侵袭、转移,该过程是一个复杂的过程,关于其在口腔鳞癌中的研究相对较少。我们在口腔鳞癌细胞系中模拟炎症微环境,探究信号通路抑制剂作用前后关键因子的表达。观察PI3K/AKT信号通路在调控转录因子实施snail的表达诱导发生EMT中的作用,探讨口腔鳞癌发生侵袭转移的相关分子机制,寻找新的诊断标记物,并为实施新的治疗策略提供有力的理论依据。
1 材料与方法
1.1 材料 人鳞癌HN4、HN6、CAL27细胞(ATCC公司);DMEM-F12培养基(GIBCO公司,美国)、TNF-α(Peprotech公司,美国)、RealMaster Mix RT-PCR 试剂盒(天根,北京)、PI3K/AKT信号通路抑制剂(LY294002)、鼠抗AKT(碧云天,上海)、VS-840-KU超净工作台(净化设备厂,苏州)。
1.2 方法
1.2.1 细胞培养及传代 人鳞癌HN4、HN6、CAL27细胞于DMEM-F12完全培养液常规培养。细胞生长至80%~90%时,用0.25%胰蛋白酶消化铺板,培养一段时间待达到一定密度且细胞状态良好时,更换无血清培养液12 h,使细胞同步化生长。
1.2.2 HN4、HN6、CAL27口腔鳞癌细胞处理 本实验主要分为以下3组:对照组、TNF-α组、LY294002组。对照组:未加TNF-α、LY294002的HN4、HN6、CAL27口腔鳞癌细胞;TNF-α组:浓度为10 μg/L的TNF-α分别刺激HN4、HN6、CAL27;LY294002组:浓度为20 μmol/L的LY294002处理细胞,1 h后加入浓度为10 μg/L的TNF-α继续刺激。
1.2.3 蛋白质免疫印迹法(western blot) 取TNF-α组、LY294002组细胞分别作用一段时间,并取相应对照组同时加入蛋白裂解液和PMSF提取细胞总蛋白。用10%SDS-PAGE凝胶将蛋白进行电泳分离,湿法转印至NC膜上,转膜结束后5%牛奶室温封闭1 h,加入AKT、P-AKT(Ser473)一抗4 ℃孵育过夜,二抗室温孵育1.5 h,加显影液后放入化学发光分析仪中曝光获取图像信息,Image lab软件扫描获取灰度值,校正内参,分析蛋白质表达情况。
1.2.4 实时荧光定量PCR(real-time PCR) 取72 h的对照组、TNF-α组、LY294002组的细胞,采用Trizol试剂提取各细胞样本的总RNA,将总RNA逆转录为cDNA后使用7500ABI real-time PCR仪,选用β-actin作为内参基因,目的基因引物序列如表1所示,上述过程均按试剂盒说明书步骤进行。采用2-△△CT相对定量法进行组间基因差异表达比较,获取数据。
1.3 统计学分析 实验中所有数据均使用Graphpad Prism 6软件进行统计分析并绘图,至少重复10次,单因素方差分析多组间差异,P<0.05为差异有统计学意义。

表1 实时荧光定量RT-PCR所需引物序列
2 结果
2.1 TNF-α作用前后、LY294002抑制剂作用后口腔鳞癌细胞形态学变化(图1) TNF-α作用72 h前后口腔鳞癌细胞HN4、HN6、CAL27形态学变化。典型的鳞状上皮细胞形态发生改变,细胞间连接也变为松散,间距拉大,形成长梭状,与成纤维细胞形态相似,提示TNF-α作用于口腔鳞癌细胞后,可能逐渐发生了EMT的形态学改变。
抑制剂(LY294002)与TNF-α联合作用72 h后口腔鳞癌细胞HN4、HN6、CAL27形态学表现为大多数细胞仍可见典型的上皮细胞形态,细胞拉长不明显,细胞间排列也较为紧密,提示LY294002处理HN4、HN6、CAL27细胞后,口腔鳞癌细胞的EMT发生可能受到一定程度的抑制。

2.2 蛋白质免疫印迹法(western blot)检测TNF-α、LY294002作用于口腔鳞癌细胞,AKT、P-AKT(Ser473)的蛋白表达 见图2~4。 TNF-α分别刺激HN4、HN6、CAL27口腔鳞癌细胞6、12、24、48、72 h后与相应对照组同时收样,每组实验各重复10次,检测TNF-α作用于口腔鳞癌细胞后AKT、P-AKT的蛋白表达(图2A、3A、4A),结果如下:发现TNF-α作用后,PI3K/AKT信号通路中关键因子AKT蛋白表达上调,且具有时间依赖性(图2B、3B、4B),HN4(F=25.28,P<0.01)、HN6(F=7.937,P<0.01)、CAL27(F=7.267,P<0.01)差异有统计学意义;P-AKT(Ser473)无明显变化趋势,差异无统计学意义,P>0.05。
在上述实验基础上,用LY294002处理HN4、HN6、CAL27细胞,1 h后加入TNF-α继续刺激6、12、24、48、72 h后与相应对照组,每组实验各重复10次,通过western blot检测AKT的蛋白表达(图2A、3A、4A),结果如下:可见LY294002作用后,AKT表达无明显变化趋势,HN4(F=0.3763,P>0.05)、HN6(F=0.1768,P>0.05)、CAL27(F=0.709 2,P>0.05)差异无统计学意义,P>0.05(图2B、3B、4B)。
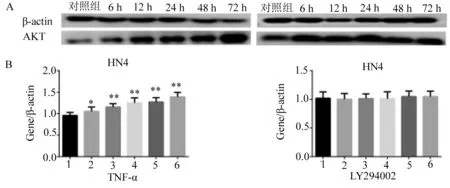
1.对照组;2.6 h;3.12 h;4.24 h;5.48 h;6.72 h;* P<0.05,** P<0.0001。
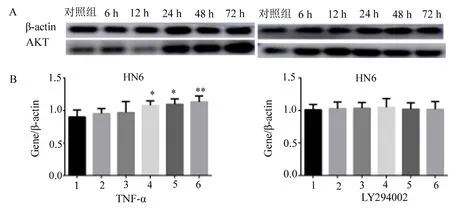
1.对照组;2.6 h;3.12 h;4.24 h;5.48 h;6.72 h;* P<0.001,** P<0.0001。
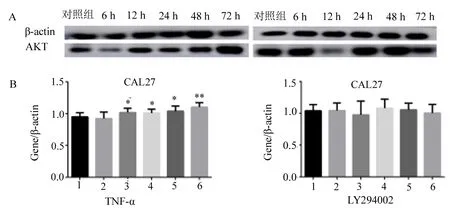
1.对照组;2.6 h;3.12 h;4.24 h;5.48 h;6.72 h;* P<0.05,** P<0.0001。
2.3 实时荧光定量PCR检测E-钙黏蛋白表达情况 在western blot基础上,TNF-α刺激口腔鳞癌细胞HN4、HN6、CAL27细胞72 h,结果显示E-钙黏蛋白的表达低于对照组,HN4、HN6、CAL27中分别是相应对照组的0.190倍(P<0.01)、0.182倍(P<0.01)、0.366倍(P<0.01),提示TNF-α刺激后诱导细胞发生EMT;而用 LY294002抑制剂处理口腔鳞癌细胞后,E-钙黏蛋白表达均较单纯TNF-α刺激升高,HN4、HN6、CAL27比TNF-α组分别相对升高1.70倍(P<0.01)、2.30倍(P<0.01)、1.30倍(P<0.01);而低于对照组,HN4、HN6、CAL27中分别是对照组的0.325倍(P<0.01)、0.418倍(P<0.01)、0.476倍(P<0.01),提示通路特异性抑制剂作用后,EMT的发生受到一定程度的抑制,说明抑制PI3K/AKT信号通路能够在一定程度上抑制EMT的发生(图5)。

图5 实时荧光定量PCR检测TNF-α以及抑制剂作用前后HN4、HN6、CAL27细胞中E-钙黏蛋白的表达
3 讨论
浸润性生长和发生远处转移是导致口腔鳞状细胞癌生存率低的主要原因,EMT现象的发生是鳞癌侵袭和转移的关键一步,它使得上皮细胞获得间质表型或成纤维样特性,其主要标志是上皮标志物E-钙黏蛋白缺失[6]。研究证实肿瘤微环境中持续低剂量的TNF-α可以通过上调转录因子snail的表达,促进口腔鳞癌细胞发生EMT[4]。当肿瘤细胞发生EMT时,其由上皮细胞向间充质细胞转化,此时上皮标志物丢失,即E-钙黏蛋白的表达会降低,相应的间充质标志物表达上升,这种变化会使细胞丧失极性,获得间质表型,形态拉长似成纤维细胞,细胞的黏附力下降,易于突破基底膜向下浸润,向周围及远处转移。实验中,我们发现在TNF-α作用后,典型的鳞状上皮细胞即铺路石样形态发生变化,表现为成纤维样特性,形态细长,且细胞间的连接减少,空隙增大;我们通过RT-PCR检测发现在TNF-α作用后,E-钙黏蛋白的表达明显降低,提示肿瘤细胞可能发生EMT。
AKT是PI3K/AKT信号通路的中心调节效应分子。通路中PI3K和mTOR可分别调节Thr308和Ser473这两个AKT的磷酸化位点(P-AKT),当两位点均磷酸化后,则可致AKT被完全活化,继而激活mTOR,使其下游靶分子4EBP1和p70S6K介导肿瘤细胞蛋白合成增加[7]。因此AKT是连接细胞存活、增殖、凋亡和细胞代谢途径的重要节点[8]。本实验在前期研究的基础上[9-10],外源性TNF-α作用于体外培养的口腔鳞癌细胞,western blot检测PI3K/AKT信号通路中上述关键因子表达,结果显示AKT蛋白表达明显升高,且具有时间依赖性。说明TNF-α刺激后,PI3K/AKT信号通路被激活,关键因子AKT表达上升。
为了进一步验证口腔鳞癌细胞侵袭转移的发生与PI3K/AKT信号通路的关系,我们再应用通路特异性抑制剂LY294002与TNF-α联合处理。发现细胞形态与对照组相似,基本保留上皮细胞形态;利用western blot检测抑制剂作用于口腔鳞癌细胞后,发现AKT的蛋白表达与对照组相似,无明显变化趋势,表明通路抑制剂作用后,AKT表达下降,PI3K/AKT信号通路在一定程度上被抑制。同时我们通过RT-PCR发现,E-钙黏蛋白的表达均升高,即LY294002作用于口腔鳞癌细胞后,通过PI3K/AKT信号通路抑制了炎症因子TNF-α通过调节snail促进EMT发生的能力,说明抑制PI3K/AKT信号通路可降低口腔鳞癌侵袭转移的作用。但E-钙黏蛋白的表达较对照组仍降低,这可能是由于该基因表达同时受多个信号通路调控,可能有其他信号通路参与所致,如由于在功能上MAPK与PI3K/AKT信号通路十分相似,因为研究表明他们之间存在一定程度的联系。人们发现MAPK的活性可以被PI3K的抑制剂减弱,比如在几内亚猪嗜中性粒细胞中,可以部分抑制血小板活化因子诱导激活MAPK的活性;wortmannin(一种PI3K的P110亚基的特异性抑制剂)会阻断CD3抗体诱导的T细胞ERK2的活化及在兔骨骼肌中MAPK的活化。Rane等分别在SB203580(MAPK通路信号抑制剂)、wortmannin、LY294002存在和不存在的条件下检测甲酰甲硫氨酰-亮氨酰-苯丙氨酸刺激的嗜中性粒细胞中的AKT Ser473的磷酸化水平,最终结果发现,SB203580、wortmannin、LY294002可以抑制AKT磷酸化,说明PI3K和MAPK均可以调节AKT的活性[11]。或者也可能是通路中存在负反馈调节机制,以上这些都仍待我们进一步研究。
综上所述,本研究显示TNF-α可通过上调PI3K/AKT信号通路中关键因子,增强口腔鳞癌细胞侵袭转移的能力,而通路特异性抑制剂作用后,TNF-α促进口腔鳞癌细胞侵袭转移的能力明显受到抑制,表明PI3K/AKT信号通路与口腔鳞癌侵袭转移的作用密切相关。我们在研究中也发现,肿瘤细胞的侵袭转移并非单一通路所调控,与其他通路之间的具体相互关系还有待进一步研究,只有对这些通路更为彻底的研究,并在此基础上采取特定的措施,才有望完全抑制肿瘤细胞侵袭转移能力。
猜你喜欢
天津医科大学学报(2021年4期)2021-08-21
云南医药(2021年3期)2021-07-21
现代临床医学(2021年2期)2021-03-29
现代临床医学(2021年2期)2021-03-29
奥秘(创新大赛)(2019年9期)2019-10-09
奥秘(2017年5期)2017-07-05
安徽医科大学学报(2015年9期)2015-12-16
医学研究杂志(2015年9期)2015-07-01
医学研究杂志(2015年12期)2015-06-10
癌变·畸变·突变(2015年3期)2015-02-27
