
牙周病的表观遗传学分析:生物信息学研究
2019-02-27 09:05王田田刘香琼李思敏
现代口腔医学杂志 2019年1期
王田田 刘香琼 李思敏
牙周炎是一种主要由菌斑生物膜导致的多因素疾病,该病可导致牙齿支持组织的进行性破坏[1]。遗传和表观遗传学因素可通过调节牙周致病菌引起的宿主免疫反应,影响牙周炎的进展[2]。为阐明牙周炎的病因机制,确定牙周炎发病过程中遗传和表观遗传因素的改变显得非常必要。
非编码RNA,作为表观遗传学的重要调控因素,有两种主要类型,微小RNA(miroRNAs,miRNAs)和长链非编码RNA(long noncoding RNAs,lncRNAs)。长链非编码RNA,微小RNA,可与信使RNA,三者之间相互作用,形成调节性竞争性内源性RNA网络[3]。微小RNA含有22个碱基对,能够负向调控信使RNA的表达[4]。最近使用芯片分析的研究[5,6]表明,牙周炎患者的炎症牙龈组织和健康牙龈组织之间在微小RNA的表达谱上存在很大差异。长链非编码RNA,作为非编码RNA的一种,包含200个以上的碱基对,可以通过正向或者负向的方式来调控编码蛋白质的信使RNA的表达[7]。研究表明长链非编码RNA在慢性牙周炎患者的炎症牙龈组织中差异性表达[8]。
应用RNA芯片分析方法研究各种RNA在牙周炎中的表达,所得到的结果存在很大的异质性[9]。因此,有必要通过生物信息学的技术,将这些既往研究中的所有的芯片数据集进行整合,从而预测出重要的与牙周炎发病风险有关的生物学标记物。近些年来,已有生物信息学研究[10]确定了与牙周炎发病过程相关的重要的基因、蛋白质,以及生物学通路。然而,基于竞争性内源性RNA理论,对牙周炎发病过程中竞争性内源性RNA网络的研究尚无报道。
本研究采用生物信息学方法,基于牙周炎的高通量RNA测序及芯片数据,通过整合长链非编码RNA,微小RNA以及信使RNA的表达谱数据,旨在构建参与牙周炎发病机制的竞争性内源性RNA(ceRNA)网络。
资料和方法
数据采集与差异表达分析:从基因表达数据库(GEO)中下载与牙周炎样品匹配的基因表达数据和miRNA表达谱数据。将炎症性牙龈样品作为实验组,健康牙龈样品作为对照组。使用R语言的limma包中的Linear模型,对牙周炎样本和牙周健康样本之间差异表达的mRNA和miRNA进行差异表达分析。P值<0.05,FDR<0.05,|log2FC(倍数变化)|≥0.58的mRNA和miRNA分别被认为是显著差异表达的基因(DEG),以及显著差异表达的miRNA(DE-miRNA)。
1.RNA互作对数据的提取
从TarBase v7.0,miRTarBase Release 6.0,和miRecords三个数据集,提取已获得实验验证的324507对miRNA-mRNA互作对。从miRcode和starBase database v2.0数据集,提取出1756对lncRNA-miRNA互作对,整合后得到DE-miRNA的mRNA靶标。
2.差异表达基因的共表达分析
为构建共表达网络和确定潜在的风险基因,利用R语言进行加权基因共表达网络分析。首先,获得P值小于0.05的显著差异表达基因的表达谱,并对基因之间的相互关系的邻接矩阵进行计算。基于公式(1),对基因互作对间的连接系数进行计算。
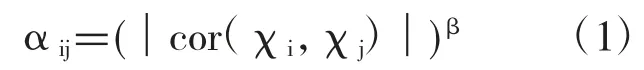
χi,χj:基因i和基因j的表达载体;cor:两个基因载体的皮尔逊相关系数;β:皮尔逊相关系数的指数变换,将皮尔逊相关系数进一步覆盖到连接系数αij。
基于网络的拓扑性质,通过加权基因共表达网络分析,获得共表达网络的模块。该网络分析了两种互作的基因以及与其互作的其他基因之间的关系。根据以下公式(2),这两种互作的基因将连接系数αij覆盖到加权系数wij上。基于加权系数,获得了显著的基因模块。

wij:代表与基因i相互作用的基因,与基因j相互作用的基因两者之间的重复。
3.共表达网络中风险基因模块的确定
为了筛选出特定的基因模块,构建了评分分类器。对每个模块进行差异表达分析,分析了在牙周炎标本和牙周健康标本之间差异表达的每个基因的表达水平,P值被设定为整合表达评分。此外,我们还计算了模块中每个基因的度(degree)与其-log10(P值)之间的关系。之后,可以筛选出明显富集的基因模块。
4.牙周炎ceRNA网络的构建和分析
从TarBase,miRTarBase和miRecords三个数据库中获取到差异表达的9498对mRNA-miRNA相互作用关系对。从miRcode和starBase v2.0两个数据库中下载了1726对DEmiRNA-lncRNA的互作对。通 过 将DEG-mRNA-miRNA互 作 对 和DEmiRNA-lncRNA互作对整合到一起,构建了竞争性内源性RNA(ceRNA)网络。
5.分析结果预测精确度的评估
为了评估本研究中分析方法的预测精确度,我们应用了含有核函数的支持向量机(SVM)。通过计算受试者工作特征(ROC)曲线(AUC)下的面积,预测模型的诊断能力得以评估。通过使用R包中的ROCR.61,整合假阳性率和敏感度的数据,绘制出ROC曲线。此外,为了可视化地显示分类器的性能,我们进行了无监督的分层聚类分析。通过将完整的联结和皮尔逊相关性作为距离测量,实现表达谱的分层聚类。
结 果
1.差异表达的mRNA,miRNAs和lncRNAs的确定
通过进行差异表达分析,我们确定了704个差异表达基因和47个差异表达的miRNA。通过使用TarBase,miRTarBase和miRecords这三个数据库,我们获得了3282对差异表达的miRNA与其靶基因的互作对,这些互作对是由37个差异表达的miRNA和2650个靶基因构成的。
2.共表达分析
通过差异表达分析,获得P值<0.05的基因并对其进行共表达分析。为了建立无标度网络,我们选择了相邻矩阵(β)的权重,此外我们还对不同β值(范围:1-20)的基因的相邻矩阵的相关系数和平均值进行了计算。当软权重为12时,无标度R∧2趋于稳定。
3.显著模块的确定
在核查已构建网络的参数后,通过对加权共表达网络进行建模,将基因分为不同模块。从加权关联网路中,检索出11个具有不同剪切静态颜色的基因模块。随机抽取出100个基因之后,对所有基因模块的基因相关性进行计算。每个基因的度k和-log10(p)之间的相关性被加权,加权结果显示这四个模块(绿色,紫色,蓝绿色,黄色)中的基因与牙周炎的发病机制显著相关(P<0.05,图1b-e)。
4.ceRNA网络的构建
在所有11个模块(图1a)中,绿色和蓝绿色模块的显著性最高,因此被用于构建ceRNA网络(图2a,b)。绿色模块包含2种DE-miRNA(miR-125a-3p和miR-142-3p)和三种DEGs(HSPA4L,EVI2B和PANK3)(图2a)。蓝绿色模块含有3种DE-miRNAs(miR-142-3p,miR-125a-3p和miR-200a),和三种DEGs(YOD1,ITGAL,和CTNNBIP1)(图2b)。在ceRNAs网络中,3种lncRNA(TUG1,FGD5-AS1,MALAT1)能够同时直接靶向两种微小RNA,包括miR-125a-3p和miR-142-3p。没有一种lncRNA能够同时直接靶向miR-125a-3p和miR-200a。
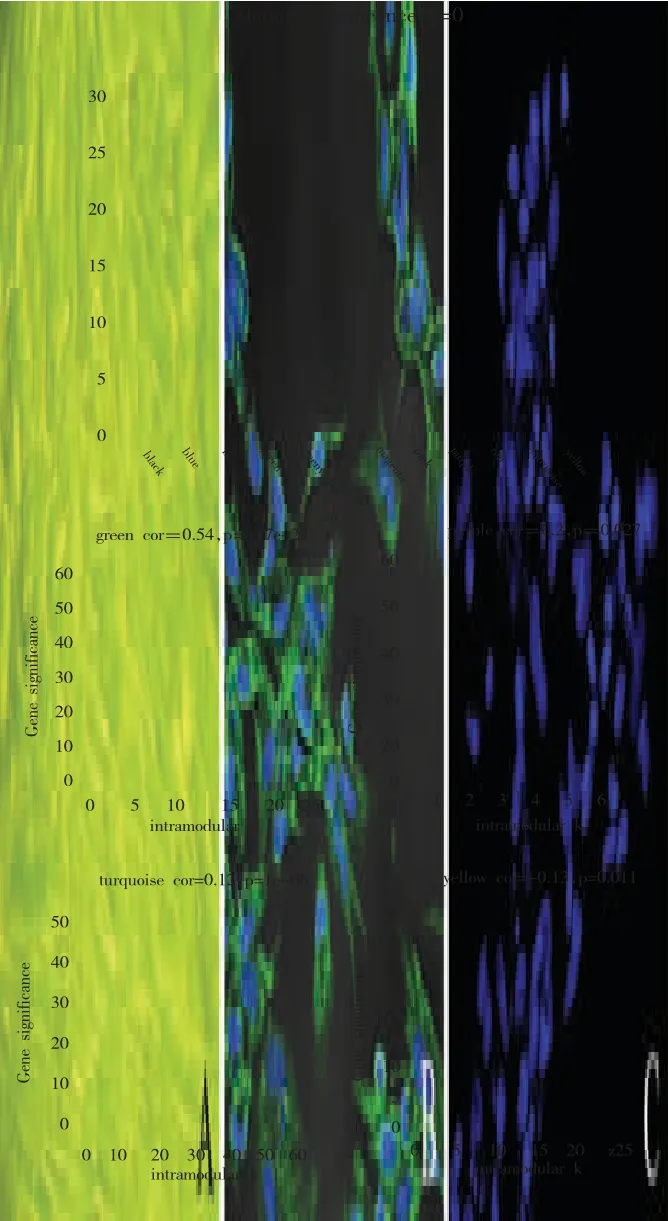
图1 共表达网络中的模块

图2 绿色模块(a)和蓝绿色模块(b)的竞争性内源性网络
5.使用两种额外的独立队列进行进一步的基因验证
为了独立验证我们预测出来的这6种基因的诊断能力,基于GPL6244这一平台,我们在GSE33774这一数据集(含有7个牙周炎样品和8个牙周健康对照样本)中,对这些基因的鉴别性能进行了检测。基于这六种基因的表达谱,我们进行了SVM分析。分析结果(图3)显示,HSPA4L基因(被hsa-miR-125a-3p所靶向)的ROC为0.9643,是这6个基因ROC中最高的。这表明,HSPA4L是在牙周炎样本与牙周健康对照样本中最为显著性差异表达的基因。

图3 ROC曲线(a),预测出的六种风险基因的热图(b)
讨 论
本研究围绕三种类型的RNA(信使RNA,微小RNA,长链非编码RNA),对牙周炎的遗传和表观遗传学机制进行剖析。六种信使RNA(HSPA4L,CTNNBIP1,ITGAL,YOD1,PANK3,和EVI2B)被 预测为牙周炎的生物标记物。HSPA4L,热冲击蛋白家族的一员,在牙周炎发病机制中发挥双重作用:一方面可以维持牙龈健康,防止受到口腔微生物的破坏[11];另一方面,HSPA在某种条件下会引发牙龈炎,加重后向牙周炎转化,进而增加牙槽骨的损失[12]。CTNNBIP1,又名β-链互作蛋白1,可抑制核内β-链蛋白的转录能力[13]。核内β-链蛋白的激活能够通过抑制非经典的Wnt通路,促进牙周韧带干细胞的增殖,并导致其成骨分化的减弱[14]。ITGAL,整合素亚基αL,可编码整合素淋巴细胞功能相关抗 原-1(LFA-1/CD11a)的 整 合 素αL链[15]。LFA-1/CD11a是一种重要的免疫调节细胞因子,可通过介导T细胞的召集和保留,调节牙周炎的免疫过程[16]。YOD1,泛蛋白酵素,在牙周炎的样本中表达下调,可被miR-30b所靶向。MiR-30家族能够负向地调控骨髓间充质干细胞的骨形态蛋白-2引起的成骨分化[17]。PANK3,泛酰酸激酶3,已被证明可被miR-142-3p所靶向;MiR-142-3p能够调节脂多糖引起的牙周炎症[9]。EVI2B,同向性病毒结合位点2B,已被证明在多种微生物感染的大鼠骨模型中表达量增加[18]。
三种微小RNA(miR-125a-3p,miR-200a,和miR-142-3p)被预测可作为牙周炎的风险生物标记物。miR-125a-3p来源于pre-miR-125a的3'末端,在牙周炎样本中表达上调。MiR-125a的上调可通过调节TRAF6/NFATc1/miR-125a调节反馈回路,抑制破骨细胞的生成,进而抑制牙周炎的进展[19]。miRNA-200a通过减少牙槽骨骨髓基质细胞(BMSCs)的成骨分化,从而抑制牙周骨再生[20]。MiR-142-3p是介导破骨细胞死亡的RANKL的独立诱导物,可通过调节Wnt通路促进成骨细胞的分化[21]。
三种长链非编码RNA(MALAT1,TUG1,和FGD5-AS1)被预测可作为牙周炎的风险生物标记物。MALAT1,肺腺癌转移相关转录子1,能够与转录因子NF-κB相互作用,调节脂多糖引发的炎症反应[22]。NF-κB信号通路可以调节RANK配体引起的破骨细胞生成,进而引起牙周炎的骨吸收[23]。TUG1,牛磺酸上调基因1,能够通过促进转化生长因子TGF-β,从而抑制细胞的增殖[24]。TGF-β信号通路在牙周炎的发病过程中,参与了牙龈成纤维细胞与牙龈上皮细胞之间的相互作用[25]。FGD5已被证明能够在牙周炎症进展过程中,通过调节血管内皮生长因子VEGF的促血管生成作用,来促进血管网络的延伸[26]。
结论:六种mRNA(HSPA4L,PANK3,YOD1,CTNNBIP1,EVI2B,和ITGAL),三 种miRNA(miR-125a-3p,miR-200a,和miR-142-3p),三种lncRNA(MALAT1,TUG1,和FGD5-AS1)参与了牙周炎的竞争性内源性RNA网络。这些结果有助于解释牙周炎的发病机制,为未来研究提供了潜在的治疗靶标。
猜你喜欢
安徽农业科学(2022年1期)2022-02-14
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年4期)2021-07-23
昆明医科大学学报(2020年12期)2021-01-26
科学(2020年3期)2020-11-26
心理学报(2020年7期)2020-07-13
中国学术期刊文摘(2016年1期)2016-02-13
中国学术期刊文摘(2016年1期)2016-02-13
中国病理生理杂志(2015年8期)2015-12-21
中国塑料(2015年10期)2015-10-14
