
低氧损伤下调大鼠脑血管内皮细胞中硫化氢及其介导的RhoA-ROCK通路激活
2019-02-22 00:44王良芳陈志武
安徽医科大学学报 2019年1期
王良芳,陈志武
缺血性损伤脑血管疾病是全球性的重大公共卫生问题,研究脑缺血损伤的机制一直是临床和基础研究较为关注的课题之一。内皮源性硫化氢(hydrogen sulfide, H2S)和一氧化氮(nitric oxide, NO)均可由脑血管内皮细胞产生和释放并参与缺血性脑损伤过程[1-3]。有研究[4-5]表明缺血性脑损伤可激活RhoA-ROCK(Ras homolog gene family, member A/Rho associated coiled coil-forming kinase)通路,并且内皮源性NO可抑制RhoA-ROCK通路的活性,但迄今尚不清楚缺血性脑损伤中,内皮源性H2S、NO及RhoA-ROCK通路三者中,孰是最先改变的原发因素以及内皮源性H2S是否也可抑制RhoA-ROCK通路的活性。因此,该文观察了大鼠脑血管内皮细胞中内皮源性H2S、NO及RhoA-ROCK通路三者在低氧性损伤中的动态变化,并探讨了内皮源性H2S对RhoA-ROCK通路活性的影响。
1 材料与方法
1.1主要试剂DMEM培养基、内皮细胞生长添加剂、胶原酶Ⅱ购自美国Sigma公司; CCK-8试剂盒、NO检测试剂盒购自南京建成生物工程研究所;RhoA活性测定试剂盒购自美国骨架细胞公司;胎牛血清、RIPA裂解液购自上海碧云天生物技术有限公司;小鼠抗β-actin多克隆抗体购自北京中杉金桥生物技术有限公司;Anti-Von Willebrand Factor antibody、兔抗ROCK1和ROCK2抗体、eNOS抗体、CSE抗体购自美国Abcam公司。
1.2方法
1.2.1大鼠脑血管内皮细胞原代培养实验动物 SD大鼠,雌雄各半,200~300 g,饲养温度在(22±2)℃,通风良好,可自由摄水进食。取大鼠5只,10%水合氯醛麻醉后置75%乙醇浸泡3 min。将大鼠置无菌操作台上,心脏灌流无菌PBS,直至肝脏和舌头发白。无菌状态下迅速断头取脑,显微镜下小心剥离脑血管后转移至超净台中预先加有胶原酶Ⅱ(工作浓度为2 mg/ml)的EP管中,用眼科镊将脑血管反复剪碎后37 ℃水浴消化30 min,之后离心弃上清液,加入含内皮细胞生长添加剂和20%新生牛血清的DMEM培养液重悬后接种于培养皿,CO2恒温恒湿孵育箱内培养,24 h后换液去除未贴壁和坏死细胞,之后视细胞生长状态换液;待80%细胞融合时,选取生长状态良好的细胞进行传代,之后按要求分组实验。
1.2.2免疫荧光染色检测培养细胞vWF表达 倒置显微镜下进行细胞形态学观察;vWF免疫荧光标记染色:取第二代状态良好的内皮细胞,胰酶消化后接种于放有载玻片的培养皿中;待细胞爬满载玻片 80%时,吸弃细胞培养液;用预冷PBS清洗3次,加入预冷的4%多聚甲醛4 ℃固定30 min;PBS 清洗3次,0.1% Triton-100室温处理10 min; PBS清洗3次,加入封闭液室温孵育1 h; 加入羊抗大鼠vWF多克隆抗体(1 ∶100) 4 ℃过夜;次日PBS清洗后,加入荧光二抗室温孵育 1 h;PBS 清洗3次,加入DAPI染色5 min,荧光封片剂封片后激光共聚焦显微镜下观察细胞染色情况并拍照。
1.2.3低氧模型制备 低氧24 h前培养基换成厌氧培养基,将细胞置于37 ℃、N295%、CO25%的可控厌氧室中。
1.2.4CCK-8法检测细胞活力 培养瓶中细胞长满后,经胰酶消化离心重悬后制成单细胞悬液,10% FBS培养液调节细胞浓度为8×104/ml,每孔100 μl接种于5块96孔板,每板设置空白对照,每组设5个复孔培养,待细胞80%融合,吸弃培养液,PBS清洗2次,加无血清培养液培养24 h后放入N295%、CO25%厌氧培养箱,分别在1、2、4、8、24 h各取出1块,并取出相应的空白对照板;向每孔加入10 μl CCK-8溶液,震荡混匀后室温下孵育1~3 h,酶标仪于450 nm波长处读取吸光度(optical density,OD)值。细胞活力(%)=(加药细胞OD-空白OD)/(对照细胞OD-空白OD)×100%。
1.2.5亚硝酸还原法检测NO含量 细胞培养液中NO代谢产物NO3在硝酸还原酶的作用下生成NO2,NO2酸性条件下与α-萘胺及对-氨基苯磺酸生成红色偶氮化合物。按说明书要求将试剂和待测样品分别加入各管后静置10 min,双蒸水调零,酶标仪于波长550 nm处测各管OD值。
1.2.6亚甲基蓝分光光度法测定H2S含量 醋酸锌可以吸收硫化氢生成硫化锌沉淀,酸性条件下硫离子可与三氯化铁 (FeCl3)和N,N-二甲基-对苯二胺硫酸盐(NDPA)反应,室温下生成稳定的亚甲基蓝,在波长670 nm处测其OD,根据标准曲线计算出H2S的浓度。
1.2.7G-LISA检测RhoA活性 按实验要求分组,待大鼠脑血管内皮细胞长满约80%后,置于冰上加冰冷裂解液提取蛋白;用蛋白质测定试剂测定蛋白质浓度,平衡各样本浓度;蛋白质提取物转移到Rho-GTP-结合蛋白预包被的平板上;400 r/min的振荡器上4 ℃孵育30 min,洗涤液常温洗2次,每次甩干;加RhoA一抗,400 r/min的振荡器上室温孵育45 min,洗涤液常温洗2次,每次甩干;加二抗振荡器400 r/min室温孵育45 min,洗涤液常温洗2次,每次甩干;每孔加HRP AB液室温下15 min后,加入HRP终止缓冲液;酶标仪在波长490 nm处读取OD值。
1.2.8Western blot检测不同低氧时间胱硫醚γ-裂解酶(CSE)、内皮型一氧化氮合成酶(eNOS)、Rho激酶ROCK1和ROCK2蛋白含量变化 分别于规定的低氧时间收集相应细胞,加入含有蛋白酶抑制剂的细胞裂解液提取蛋白。用BCA试剂盒测定总蛋白含量,各组取30 μg等量蛋白煮沸变性,行聚丙烯酰胺凝胶电泳,然后将蛋白转移到硝酸纤维素膜上,封闭后加入兔抗鼠ROCK、eNOS、CSE多克隆抗体一抗(1 ∶1 000)孵育;4 ℃过夜,次日洗膜后加入二抗室温孵育1 h,洗膜后加入显色液显影曝光内皮细胞L显色,Image J定量分析灰度值。
1.3统计学处理采用SPSS 16.0软件进行分析。两组之间独立样本进行t检验,多组之间单因素方差分析,P<0.05为差异具有统计学意义。
2 结果
2.1原代培养内皮细胞鉴定光镜下原代培养的内皮细胞为梭形或多边形,旋涡状生长,呈聚集状,贴壁牢固(图1A)。免疫荧光染色显示:细胞核DAPI染色为蓝色荧光,超过90%的细胞胞质存在红色荧光,说明内皮细胞纯度达90%(图1B)。
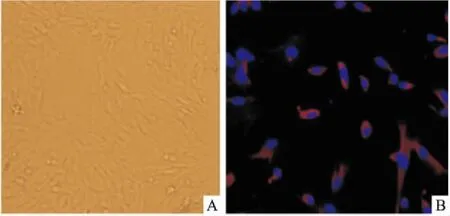
图1 原代培养的大鼠脑血管内皮细胞和免疫荧光鉴定
2.2不同低氧时间对大鼠脑血管内皮细胞活力的影响与control组比较,低氧时间延长细胞活力明显下降。低氧8 h细胞活力下降了(39.3±4.3)%,低氧24 h后细胞活力下降(60.9±6.1)%,差异均有统计学意义,表明低氧可致大鼠脑血管内皮细胞活力下降。见图2。
2.3大鼠脑血管内皮细胞低氧后NO、H2S含量及RhoA活性的动态变化大鼠脑血管内皮细胞低氧1、2、4、8、24 h后分别检测H2S、NO含量及RhoA活性变化,与control组比较,低氧1 h后H2S含量显著下降(F=4.23,P<0.01);NO含量在低氧4 h后才开始显著下降(F=3.19,P<0.05);RhoA活性低氧8 h后显著增加(F=7.31,P<0.01)。上述三者变化差距随时间延长而显著加大。见图3。因此定量结果表明在大鼠脑血管内皮细胞低氧中,H2S降低最早,NO次之,RhoA活性增加再次之。
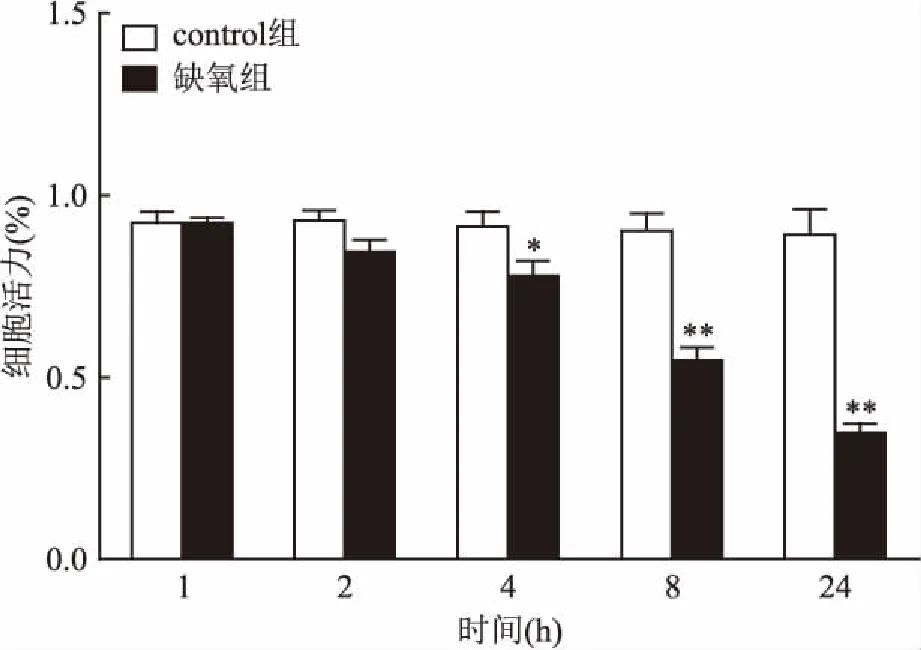
图2 不同低氧时间对大鼠脑血管细胞活力的影响
与control组比较:*P<0.05,**P<0.01
2.4低氧不同时间对大鼠脑血管内皮细胞CSE、eNOS、ROCK1和ROCK2蛋白表达的影响Western blot结果显示:CSE和eNOS蛋白随低氧时间延长呈持续下降趋势,低氧4 h时CSE蛋白表达较正常组有了显著下降;低氧8 h后eNOS蛋白才开始明显降低;相反ROCK1、ROCK2蛋白表达随低氧时间延长呈上升趋势,低氧8 h时ROCK1、ROCK2蛋白表达显著增加。见图4。上述各蛋白的变化随低氧时间延长而显著加大。
2.5H2S对大鼠脑血管内皮细胞中RhoA活性的影响
2.5.1内源性H2S对RhoA活性的影响 与control组比较,RhoA特异性抑制剂C3转移酶(C3 Transferase,C3 TF)(25 μg/ ml,8 h)可显著抑制RhoA活性,差异有统计学意义(P<0.05),但CSE抑制剂DL-炔丙基甘氨酸(DL-propargylglycine,PPG)(10 mmol/L,4 h)可显著加RhoA活性,差异有统计学意义(P<0.05),提示内源性H2S可抑制RhoA的激活。见图5。
2.5.2内源性和外源性H2S对C3转移酶诱导的RhoA活性降低的影响 与C3TF组比较,PPG组(10 mmol/L,4 h)可显著抑制C3TF诱导的RhoA活性降低,差异有统计学意义(P<0.05),提示内源性H2S均可增强C3TF诱导的RhoA活性降低。H2S供体NaHS(50 μmol/L、30 min)可直接增强C3TF诱导的RhoA活性降低,差异有统计学意义(P<0.05)。见图5。
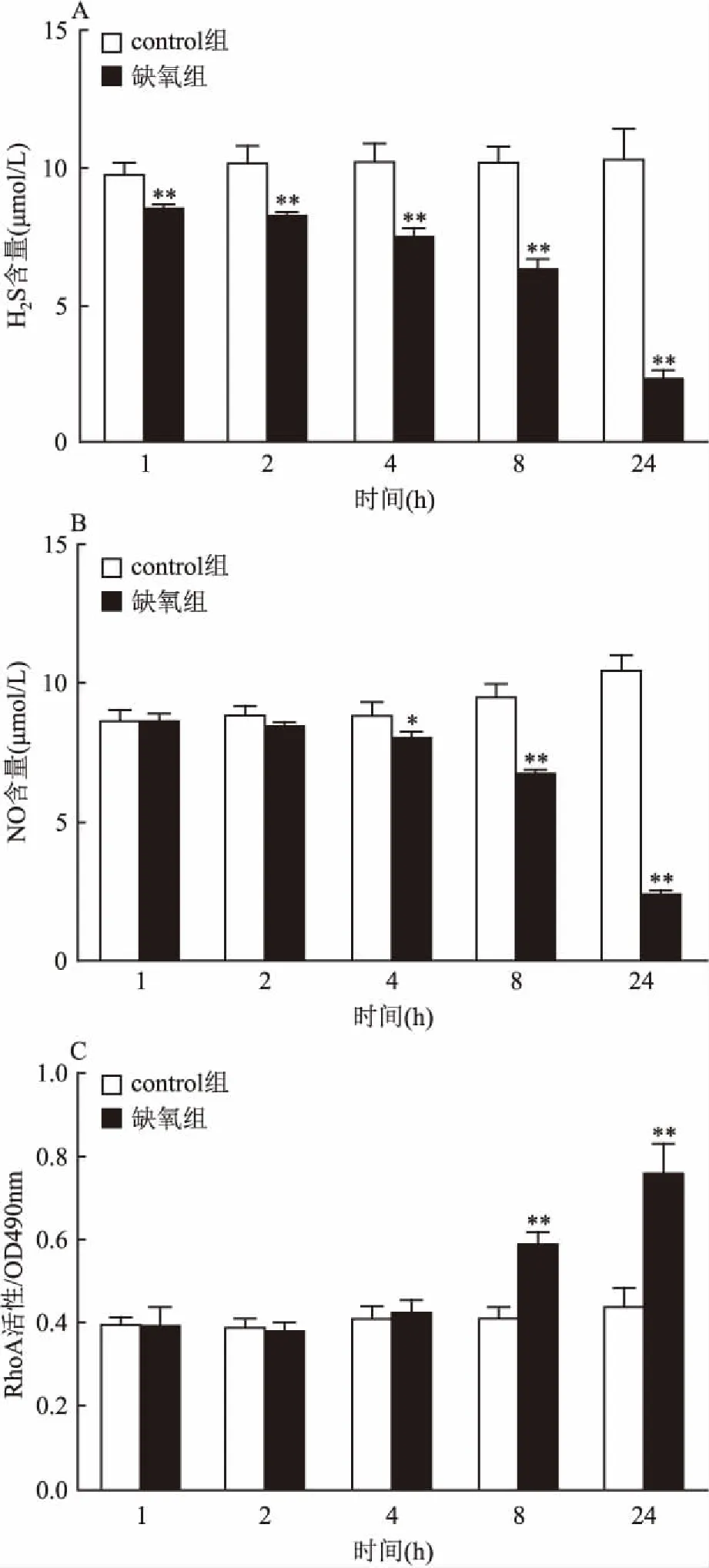
图3 大鼠脑血管内皮细胞低氧H2S、NO的含量和RhoA活性的动态变化(n=3)
A:H2S含量动态变化;B:NO含量动态变化;C:RhoA活性动态变化;与 control组比较:*P<0.05,**P<0.01
2.5.3外源性H2S对RhoA活性的影响 外源性H2S供体硫氢化钠(sodium hydrosulfide,NaHS)10、50和100 μmol/L处理30 min,与control组比较差异有统计学意义,可明显并呈一定的浓度依赖性地降低RhoA活性,提示外源性H2S也可抑制RhoA的激活。见图6。
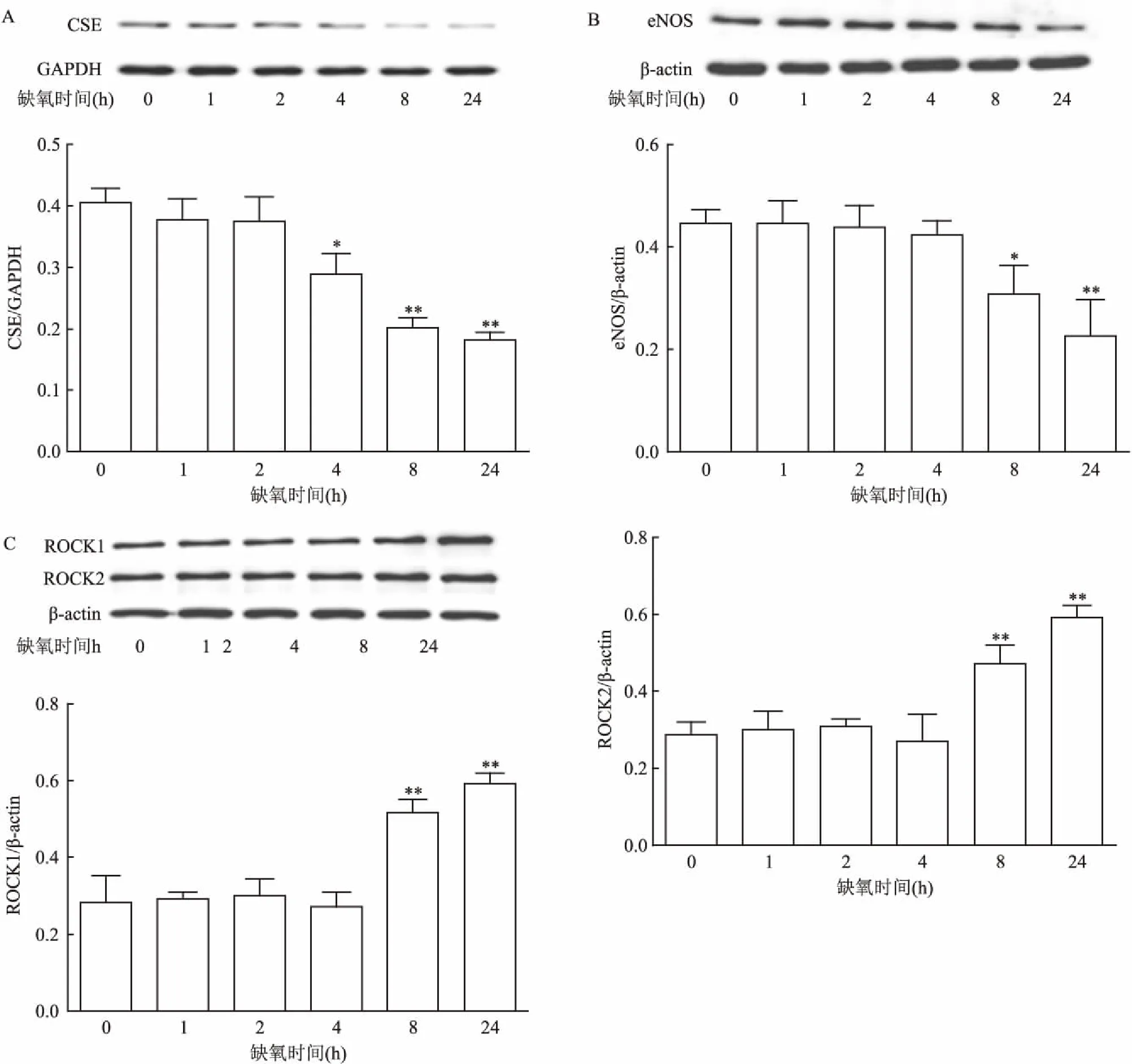
图4低氧不同时间诱导大鼠脑血管内皮细胞损伤后CSE、eNOS、ROCK1、ROCK2蛋白表达的影响(n=3)
A:CSE表达水平;B:eNOS表达水平;C:ROCK1/2表达水平;与control组比较:*P<0.05,**P<0.01
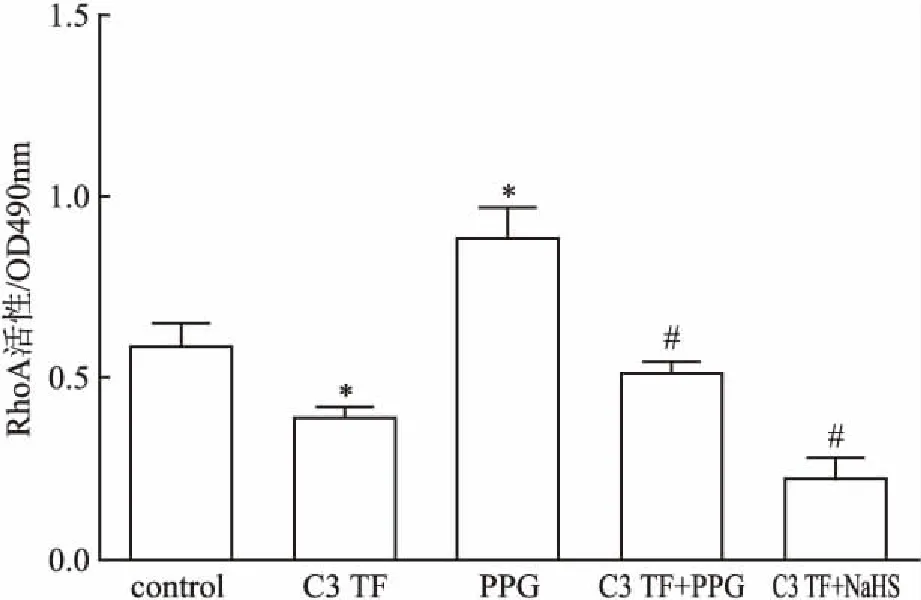
图5 PPG对大鼠脑血管内皮细胞中RhoA活性的影响(n=3)
与control组比较:*P<0.05;与C3 TF组比较:#P<0.05
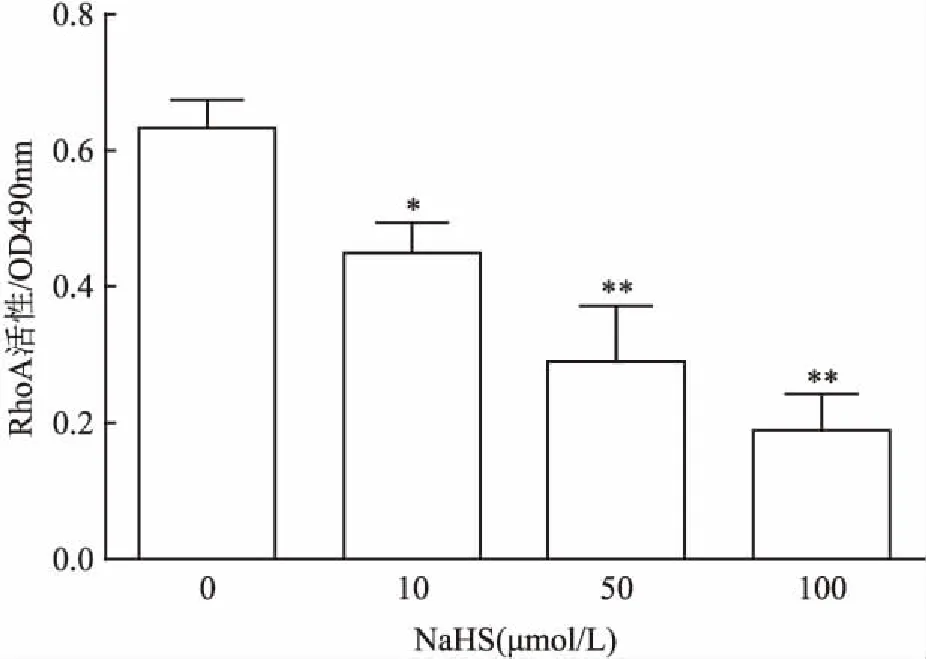
图6 NaHS对大鼠脑血管内皮细胞中RhoA活性的影响(n=3)
与control组比较:*P<0.05,**P<0.01
3 讨论
小G蛋白的Rho家族包括RhoA、Rac1和Cdc42[6-7],其中RhoA是最具特色的蛋白质,它作为分子开关在无活性的GDP结合和活性GTP结构之间循环,与下游靶点相互作用以引发各种细胞反应。研究[8]表明,RhoA-ROCK通路与心血管疾病发病机制有关,如冠状动脉粥样硬化性心脏病、高血压、心力衰竭等,其抑制剂法舒地尔(fasudil)可以治疗许多心血管疾病。
缺血性脑损伤是一严重危害人类健康的脑血管疾病,脑血管内皮与缺血性脑损伤的发生密切相关。内皮源性NO和H2S是调节脑血管内皮舒张功能的两种重要的血管内皮舒张因子,其改变参与了脑缺血损伤过程。在急性脑缺血动物模型中,有研究[9]显示脑缺血可激活ROCK,而ROCK抑制剂fasudi或Y-27632可抑制ROCK的激活,并减少脑梗死面积。因此,RhoA-ROCK通路的激活也是脑缺血损伤过程中的一个因素。本研究表明在大鼠脑血管内皮细胞低氧损伤过程中,内皮源性H2S和内皮源性NO分别在低氧1 h和4 h时发生了明显降低,而RhoA-ROCK通路活性在8 h时才有明显增加。并且Western blot检测结果也表明内皮源性H2S合成酶CSE和内皮源性合酶eNOS蛋白表达的降低及ROCK蛋白表达的增高分别发生在大鼠脑血管内皮细胞低氧的4 h、8 h和8 h时,也支持着内皮源性H2S降低发生最早。上述的实验结果还表明内皮源性H2S在低氧1 h时就明显降低了,但其合成酶CSE蛋白表达在4 h时才显著降低,提示在大鼠脑血管内皮细胞低氧过程中,H2S合成酶CSE的活性也有显著降低,并且早于其蛋白表达的下降。因此,本研究结果表明在大鼠脑血管内皮细胞低氧损伤过程中,内皮源性H2S降低发生最早,其次为NO,而RhoA-ROCK通路的激活迟于前二者。
在低氧损伤过程中,大鼠脑血管内皮中RhoA-ROCK通路改变迟于NO和H2S的降低,但前者的改变是否由NO和H2S降低引起的呢?有研究[10]表明NO可阻止RhoA从细胞质转位到细胞膜上,从而抑制RhoA的活化。本研究表明内源性H2S和外源性H2S均可抑制RhoA的激活。并且内源性和外源性H2S均可增强C3 TF诱导的RhoA活性降低,也支持着H2S可抑制RhoA的激活。因此,结合前述的内皮源性H2S降低发生在前,RhoA-ROCK通路激活发生在后,可以认为在低氧损伤过程中,大鼠脑血管内皮细胞中RhoA-ROCK通路的激活可能是继发于H2S的降低。至于内皮源性H2S与NO的相互作用已有大量文献报道,此处不再赘述了。
综上所述,在大鼠脑血管内皮细胞低氧损伤过程中,内皮源性H2S降低发生最早,其次为NO,而RhoA-ROCK通路激活发生在后,可能是继发于H2S的降低。
猜你喜欢
中国典型病例大全(2022年13期)2022-05-10
现代临床医学(2021年6期)2021-11-20
天津医科大学学报(2021年3期)2021-07-21
中国中医急症(2019年10期)2019-05-21
国外医药(抗生素分册)(2016年5期)2016-07-12
川北医学院学报(2015年5期)2015-12-05
中国医疗美容(2015年1期)2015-07-12
中国当代医药(2015年33期)2015-03-01
西南国防医药(2015年11期)2015-02-28
中国病理生理杂志(2015年10期)2015-01-26
