
Capture a live-photo of tumor infiltrating T cell landscape at single cell resolution
2019-02-16 06:06XinDongRuidongXue
Cancer Biology & Medicine 2019年4期
Xin Dong, Ruidong Xue
Translational Cancer Research Center, Peking University First Hospital, Beijing 100034, China
Immune checkpoint blockade therapy, which targets T cells to enhance the antitumor immune response, has become a promising treatment for many cancer types1,2. Till now, a total of 6 checkpoint inhibitors targeting cytotoxic T lymphocyte antigen-4 (CTLA-4), programmed death-1 (PD-1), or programmed death-ligand 1 (PD-L1) have been approved by U.S. FDA, and many more are in different stages of clinical trials3. However, therapeutic efficacy varies among different patients and cancer types, and the underlying mechanism is not fully understood4,5. Tumor-infiltrating lymphocytes (TILs) play a central role in tumor immunity and show extensive heterogeneity. On the one hand, TILs are composed of different functional subsets, such as effector T cells, regulatory T cells (Tregs), exhausted T cells (TEX), etc.6On the other hand, the heterogeneous nature of TILs also lies in the tremendously diverse T cell receptor (TCR) repertoire resulting from V(D)J loci recombination7. Therefore,comprehensive profiling of TILs in different cancer types,ideally at single cell resolution, is needed to characterize their genetic and functional heterogeneity, explain the efficacy variance of immune checkpoint therapy, and facilitate future patient stratification.
Recent advances in single-cell RNA sequencing (scRNAseq) allow us to investigate the heterogeneous TILs among different cancer types with unprecedented detail8,9. For instance, Savas et al.10identified a novel cell subset similar to tissue-resident memory cells (TRM) in triple-negative breast cancer (TNBC) and defined them as CD8+CD103+TRM-like cells. Interestingly, they found that these cells expressed high level of both effector proteins and immune checkpoint molecules. Based on these results, they proposed a CD8+TRMgene signature that was associated with a better prognosis in early-stage TNBC. Kurtulus et al.11depicted the dynamics of CD8+TILs induced by checkpoint blockade in a preclinical mouse model of colon cancer. They found that combined Tim-3+PD-1 blockade induced a shift from naive-like to memory-precursor- and effector-like subsets within PD-1–CD8+TILs. They further revealed that expression of Tcf7 was a key regulator of the memory-precursor-like cells and a requisite for the efficacy of diverse immunotherapies.
These pioneer studies characterized the heterogeneous populations of TILs in different cancer types, but the heterogeneity regarding TCR regions remains unknown. To dig into this aspect, Stubbington et al.12developed a computational tool called TraCeR to reconstruct paired αand β-chain TCR sequences from scRNA-seq data. Given the vast different combination of TCR regions, they assume that cells with identical TCR sequences are from the same clonal origin while those with different TCR sequences are all independent occurrences. Along with the expression profiles of known signature genes, scRNA-seq data were able to determine the cell identity of TILs. Hence, the TraCeR algorithm links cell identity with clonal status of each single T cell, providing a powerful tool to compare transcription profiles across different T cell subsets.
Taking advantage of full-length mRNA transcripts generated by Smart-Seq213,14and TCR sequences extracted by TraCeR (Figure 1), Zemin Zhang and his colleagues systematically interrogated TILs landscape in hepatocellular carcinoma (HCC)15, non-small cell lung cancer (NSCLC)16and colorectal cancer (CRC)17. Most importantly, much emphasis has been put on comparing TILs across matched tumor-normal-blood sample trios from the same patient(Figure 1). With this spatial information, they were able to reconstruct a live-photo of TILs landscape from the otherwise static captures of the tumor, investigate the clonal expansion, state transition, and migration of TILs, thus revealing critical new details of immune surveillance events during carcinogenesis.
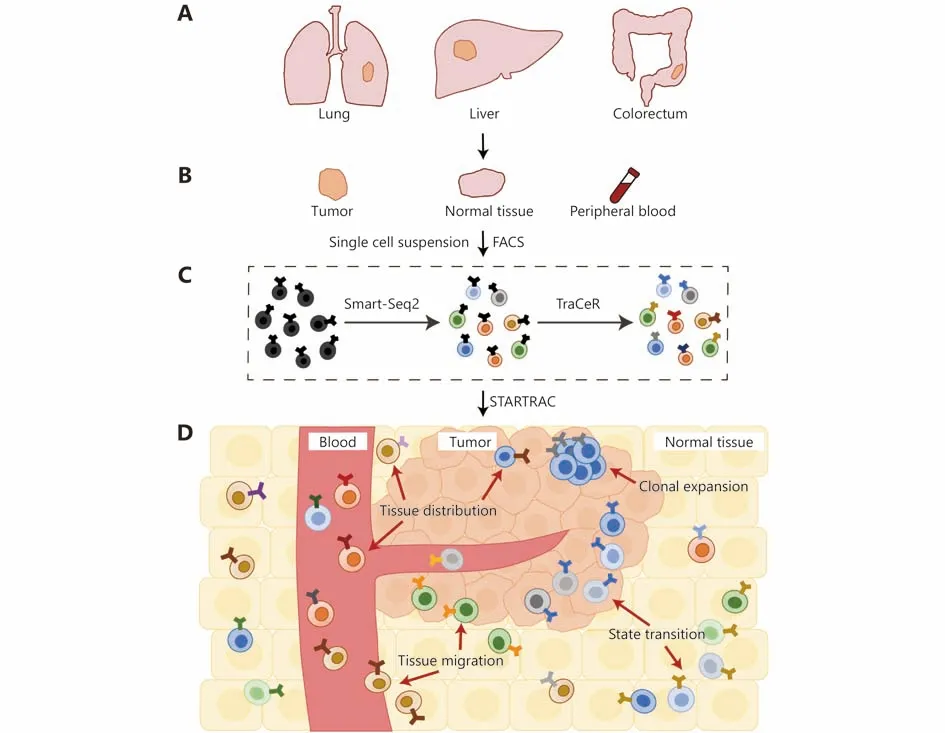
Figure 1 Reconstructing the dynamic TILs landscape with STARTRAC toolkit. (A, B) Matched tumor-normal-blood sample trios are isolated from different cancer types. Single cell suspension is made from these samples and sent for FACS to isolate T cells. (C) Full-length mRNA transcript profile of each single cell is characterized by Smart-Seq2, which is then fed to TraCeR to reconstruct its TCR sequence. (D) T cell dynamics across tumors, normal tissues and blood are analyzed by STARTRAC analysis toolkit. STARTRAC-dist depicts the tissue distribution preference of different T cell subsets. STARTRAC-expa depicts clonal expansion of T cells, which have same TCRs and cell identities.STARTRAC-migr depicts clonal T cells located in different tissues, suggesting potential migrations. STARTRAC-tran depicts T cells with identical TCRs yet exhibiting different developmental states, indicating potential state transitions.
Zheng et al.15sequenced 5,063 T cells isolated from six treatment-naive HCC patients. A total of 11 T cell subsets were identified. Compared with blood and adjacent normal tissues, a much higher percentage of clonal CD8+T cells and Tregswas observed in the tumor. Integrating pseudotime trajectory and TCR analysis, they portrayed a T cell developmental process transiting from naive to exhausted states. Besides, they identified a novel signature gene of tumor Tregsand CD8+TEX, LAYN. Notably, the LAYNoverexpressed CD8+T cells produced significantly less interferon-γ (IFNG). In line with this, higher LAYN expression was associated with poorer survival within the TCGA HCC dataset. These results indicate that LAYN expression in T cells may serve as a prognostic biomarker in HCC.
Guo et al.16sequenced 12,346 T cells isolated from 14 treatment-naive NSCLC patients and identified a total of 16 T cell subsets. They observed a high proportion of clonal effector T cells existing in both blood and solid tissues,indicating that these inter-tissue T cells have a highly migratory nature. Interestingly, they discovered two “preexhausted” T cell subsets, CD8-C4-GZMK and CD8-C5-ZNF683. Survival analysis of these signature genes in the TCGA lung adenocarcinoma (LUAD) cohort further revealed that a higher ratio of “pre-exhausted” to exhausted T cells was associated with a better prognosis. Also, spatial heterogeneity of Tregswas observed, with CD4-C8-FOXP3 and CD4-C9-CTLA4 enriched in blood and tumors,respectively. Notably, the bimodal expression of TNFRSF9 indicated the functional heterogeneity of Tregs. Further comparison of TNFRSF9+and TNFRSF9-Tregsled to the discovery of a 260-gene signature, particularly IL1R2,correlating with poor prognosis in LUAD. These results provide critical information for the immunotherapies in NSCLC.
Zhang et al.17sequenced 11,138 T cells isolated from 12 treatment-naive CRC patients, including 4 microsatelliteinstable (MSI) and 8 microsatellite-stable (MSS) cases. A total of 20 T cell subsets were identified. To portray a dynamic T cell landscape across different locations, they developed an analysis toolkit called STARTRAC (single T-cell analysis by RNA-seq and TCR tracking). It comprises four indices, -dist, -expa, -migr, and -tran, depicting the tissue distribution, clonal expansion, migration and developmental transition of T cells, respectively. With STARTRAC-dist, they showed that exhausted T cells were enriched in the tumor,and different Tregssubsets exhibited tissue preference. With STARTRAC-expa, highly clonal expansion was found in both CD8+TEXand TEMRA. Unexpectedly, CD8+TEXalso exhibited high expression level of effector molecules, such as IFNG,GZMB/GZMH and PRF1, suggesting retained antitumor effector potential. With STARTRAC-tran, they revealed a binary developmental trajectory from TEMto either TEMRAor TEX, distinct from the scenario in HCC that TEXwere evolved from TEMRA15. This TCR-based fate decision implies that promoting the transition from TEMto TEMRAis a potential way to enhance immune activity.
To dig into the difference of immune activity between MSS and MSI patients, they compared their T cell landscape and found that CXCL13+BHLHE40+TH1-like cells were significantly enriched in MSI tumors. Given that MSI patients had better response to the immune checkpoint inhibition than MSS ones18, CXCL13+BHLHE40+TH1-like cells might confer MSI patients substantial sensitivity to immunotherapies. Gene expression analysis of CXCL13+BHLHE40+TH1-like cells revealed increased expression of IGFLR1, which was also upregulated in tumor TEXand Tregs. At last, they showed that IGFLR1 could synergize with TCR signaling and serve as a co-stimulatory molecule.
Since the HCC and NSCLC datasets were generated with the same experimental strategy (Figure 1), Zhang et al.17were able to re-analyze these data with STARTRAC and compare the TILs landscape across 3 cancer types. They found that T cell patterns were distinct in both tumors and normal tissues.Interestingly, CRC and HCC tumors showed a higher abundance of CD8+TEXand CD4+Tregs, while NSCLC tumors showed enrichment of tumor TRMcells.
Besides, similar approaches combining scRNA-seq with TCR sequencing were also adopted by other groups. The key difference is that these studies focused on T cells from the tumor. For instance, using Smart-seq2 but a different TCR extraction algorithm MiXCR, Sade-Feldman et al.19sequenced 19,392 immune cells isolated from 32 metastatic melanoma patients. Importantly, these patients were treated with anti-PD1 therapy, among which 48 tumor biopsies were collected before or after the treatment. Seventeen lesions exhibited therapeutic responses while 31 lesions were nonresponding. Two major CD8+cell subsets, CD8_G and CD8_B, were identified when comparing the cellular composition of these two groups of lesions. CD8_G cells,showing increased expression of genes linked to memory,activation, and cell survival, and reduced expression of coinhibitory molecules, were enriched in responding lesions. By contrast, CD8_B cells, exhibiting high level of genes linked to cell exhaustion, were enriched in non-responding lesions.Among the top genes enriched in CD8_G cells, they identified and validated that TCF7 could predict a better outcome for checkpoint therapy. Notably, they performed TCR analysis and identified four different patterns of expansion, which were closely associated with cell states and clinical outcomes.
With MARS-seq and TCR-seq developed by their own laboratory, Li et al.20analyzed 29,825 T and NK cells from 25 melanoma patients. Notably, they adopted a novel MetaCell algorithm to identify homogeneous and robust cell subsets21.They revealed that dysfunctional CD8+T cells showed gradually increased expression of immune checkpoint genes,suggesting a gradient of T cell dysfunction. By contrast, the CD8+cytotoxic T cells were unconnected to this gradient and stood out as a distinct population. TCR analysis further revealed that proliferative CD8+T cells were highly enriched in the early stages of dysfunction. At last, they expanded T cells ex vivo and tested their reactivity towards autologous tumor digest, and found that CD8+T cells with high dysfunctionality level were correlated with better tumor reactivity.
Collectively, these studies revealed a comprehensive and dynamic landscape of TILs among multiple cancer types at single cell resolution, providing rationales for the efficacy variance of existing checkpoint therapies, implications for establishing new standard of patient stratification and potential new immune therapeutic targets for future clinical trials. Especially, the STARTRAC related experimental and analytical pipeline enabled us to capture a live-photo of the TIL landscape during tumor progression. However, much more details of the dynamic immune landscape await us to explore. Firstly, most of the above studies focused on T cells,the detailed landscape of other types of immune cells, like B cells, macrophages and dendritic cells, etc. remains unknown.Secondly, considering that intra-tumor heterogeneity is a well-characterized feature of tumor, to what extent the T cell composition differs among multiple regions in a single tumor and how this heterogeneity impacts the immunotherapies warrant further studies. At last, from the evolutionary point of view, tumorigenesis is a long-term, complicated process of different clones competing for growth advantages. How the evolving tumor clones or subclones interact with different immune cell populations remains elusive. One step further,how different immunotherapies will influence this mixed,interactive, dynamic tumor-immune landscape of different cancer types is still a mystery. More new experimental designs and bioinformatic tools are needed for us to answer these fundamental questions.
Acknowledgments
This work was supported by National Natural Science Foundation of China (Grant No. 81802813) and Scientific Research Seed Fund of Peking University First Hospital(Grant No. 2018SF039).
Conflict of interest statement
No potential conflicts of interest are disclosed.
 Cancer Biology & Medicine2019年4期
Cancer Biology & Medicine2019年4期
- Cancer Biology & Medicine的其它文章
- Clinical proteomics: a driving force for cancer therapeutic target discovery and precision medicine
- The crosstalk between autophagy and ferroptosis: what can we learn to target drug resistance in cancer?
- Technical advances in NK cell-based cellular immunotherapy
- The potential mechanism, recognition and clinical significance of tumor pseudoprogression after immunotherapy
- Crucial role of Anxa2 in cancer progression: highlights on its novel regulatory mechanism
- The challenge of drug resistance in pancreatic ductal adenocarcinoma: a current overview