
Clinical proteomics: a driving force for cancer therapeutic target discovery and precision medicine
2019-02-16 06:06:56HaoWangNaJiangYaliZhangRuibingChen
Cancer Biology & Medicine 2019年4期
Hao Wang, Na Jiang, Yali Zhang, Ruibing Chen
1Department of Genetics, School of Basic Medical Sciences; Tianjin Medical University, Tianjin 300070, China; 2 School of Pharmaceutical Science and Technology, Tianjin University, Tianjin 300072, China
Precision medicine based on the accurate and comprehensive molecular characterization of tumors has become the most promising therapeutic strategy for cancer. In current clinical practice, patients with cancer normally receive the same treatment as other patients with the same type of cancer.However, people with different molecular backgrounds may respond quite differently to the same treatment. The promise of precision medicine is that treatments may be specifically designed for individual patients when the molecular information of the tumor is available, ensuring that patients receive the treatment to which they would be most likely to respond.
With advances in next-generation sequencing (NGS)technology, genomic profiles of many different types of cancers have been generated in large-scale DNA sequencing projects, such as The Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium (ICGC). These projects have characterized a substantial number of genetic abnormalities that may be used for personalized targeted therapy. The application of genetic profiling has achieved great success and improved the survival of many patients1,2.However, response rates to targeted therapies predicted solely by genetic profiles are not optimistic, suggesting that a more comprehensive understanding of tumor biology is required to develop more effective treatment strategies.
As the ultimate executor of cellular processes, the proteome is an indispensable link in precision medicine. A series of regulatory steps, e.g., transcription, epigenetic regulation, alternative splicing, RNA processing, RNA transportation, translation, post-translational modification(PTM) and protein degradation, are involved in the pathway from genes to the expression of the proteins responsible for the phenotype. Therefore, protein levels are not solely dependent on the abundance of their template mRNAs. The profiling of mRNAs using sequencing may provide some information about gene expression, but accumulating evidence has revealed a poor correlation between the abundance of mRNAs and the levels of their protein products. An estimated 30%–70% of genes lack a statistically significant positive correlation between their mRNA and protein levels3. The ambiguous relationship between mRNAs and proteins is mainly attributed to the multiple steps involved in the mechanism regulating gene expression and the complex and unstable post-transcriptional modifications.In addition, the different methods of sample preparation and data acquisition between NGS-based transcriptomics and mass spectrometry (MS)-based proteomics may also contribute to the poor correlation between transcriptomic and proteomic data. Since majority of the drugs used in targeted therapy act on proteins, an examination of the expression and modifications of proteins is indispensable for precision oncology from drug development to the design of personalized treatment. Unfortunately, the large-scale proteomic profiling of cancers has substantially lagged behind the genomic and transcriptomic studies. In this editorial, we summarize the recent efforts in the proteomic profiling of cancers and comment on the value of clinical proteomics in different phases of precision medicine.
Large-scale proteomic profiling of tumors
In recent years, due to the advances in NGS technology and technical improvements in MS based proteomics,proteogenomics, the integration of proteomics with genomics, has become an emerging technology to more comprehensively study the molecular signature of tumors.For example, the National Cancer Institute’s Clinical Proteomic Tumor Analysis Consortium (CPTAC) that launched in 2011 is attempting to obtain a better understanding of the molecular basis of cancer by integrating both large-scale proteomic and genomic analyses. CPTAC has documented the proteomes of several types of tumors,such as colorectal, breast and ovarian tumors, using tumor tissues for which genomic profiles were previously reported by TCGA4-6. The application of large-scale proteome profiling will improve our understanding of the molecular basis of cancer that cannot be completely elucidated or is impossible to determine using genomics alone.
The general strategy for a proteogenomic analysis and its potential applications in precision medicine are shown in Figure 1. DNA and RNA are extracted from the tumor samples and analyzed using NGS to profile genomic abnormalities and gene expression. For proteomics, proteins are extracted from samples, digested into peptides and fractionated with liquid chromatography (LC) to reduce the sample complexity. Next, the pre-fractionated peptides are analyzed using shotgun proteomics with liquid chromatography-tandem mass spectrometry (LC-MS/MS).Proteins could be quantified using different approaches, such as label-free approaches or isobaric labeling7. CPTAC studies normally employ isobaric tags for relative and absolute quantification (iTRAQ) for relative protein quantification across tumor samples. Candidate protein sequences derived from exome or RNA sequencing data are used to build a customized tumor-specific protein sequence database for protein identification. Thus, sample-specific peptides that are missing from the general protein reference databases are detected, leading to the identification of tumor-specific genomic variations, such as somatic mutations, frameshifts,mutations at exon-exon junctions and gene fusions.Subsequently, the molecular profiles are integrated and correlated with clinical information to understand and predict cancer subtypes, as well as their prognostic features and potential responses to treatment strategies.
In addition to the studies conducted by CPTAC, other groups have also reported large-scale proteomic studies on different types of cancers, as summarized in Table 1. The articles discussed here were retrieved from PubMed and selected based on the following criteria: 1. the studies were performed on resected solid tumor tissues, 2. multi-omic data were integrated, 3. the sample size was larger than twenty, and 4. studies were published in the last five years. Ge et al.9studied paired tumor and none-tumor tissues from 84 patients with diffuse gastric cancer (DGC) and clustered the tumors into three subtypes based on proteomic data.Subsequently, Mun et al. reported the proteomic profiling of DGC in young populations and identified four subtypes with different prognoses from 80 patients with DGCs by performing an integrated analysis of mRNA, protein,phosphorylation, and N-glycosylation data12. This study also correlated phosphorylation and N-glycosylation data with somatic mutations, providing in-depth descriptions of the correlations between signaling pathways and genomic abnormalities. Sinha et al.13analyzed 76 patients with intermediate-risk prostate cancers by integrating genomics,epigenomics, transcriptomics and proteomics to provide comprehensive information about the molecular characteristics of prostate cancer. Archer et al.10conducted a multi-omic study of 45 patients with medulloblastoma by integrating genomics, RNA sequencing and proteome profiling, described the molecular signatures of medulloblastoma subgroups, identified distinct signaling pathways associated with the subsets of tumors, and revealed novel potential therapeutic targets in medulloblastoma.There is an ongoing effort by the global proteomic community to characterize additional cancer types and to expand the applications of clinical proteomics to obtain a better understanding of the drug responses and resistance to therapies, which will accelerate the translation of molecular profiling into the clinic.
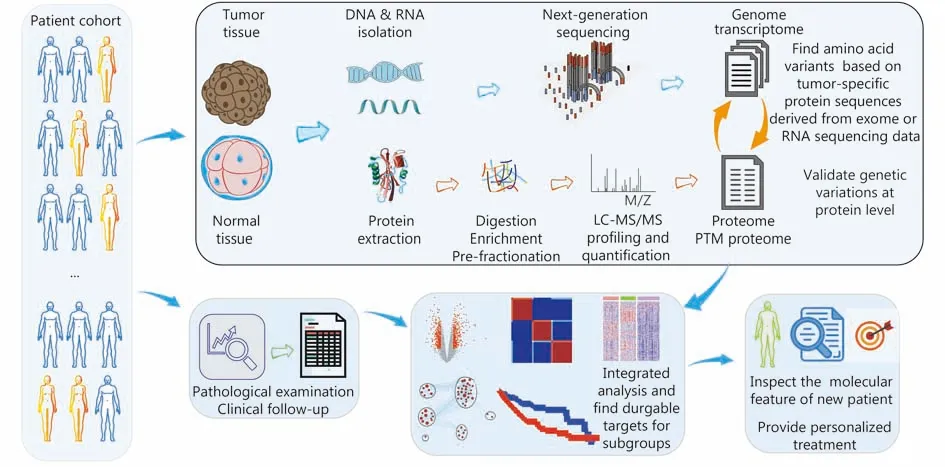
Figure 1 The application of proteogenomic analysis in the precision medicine of cancer.
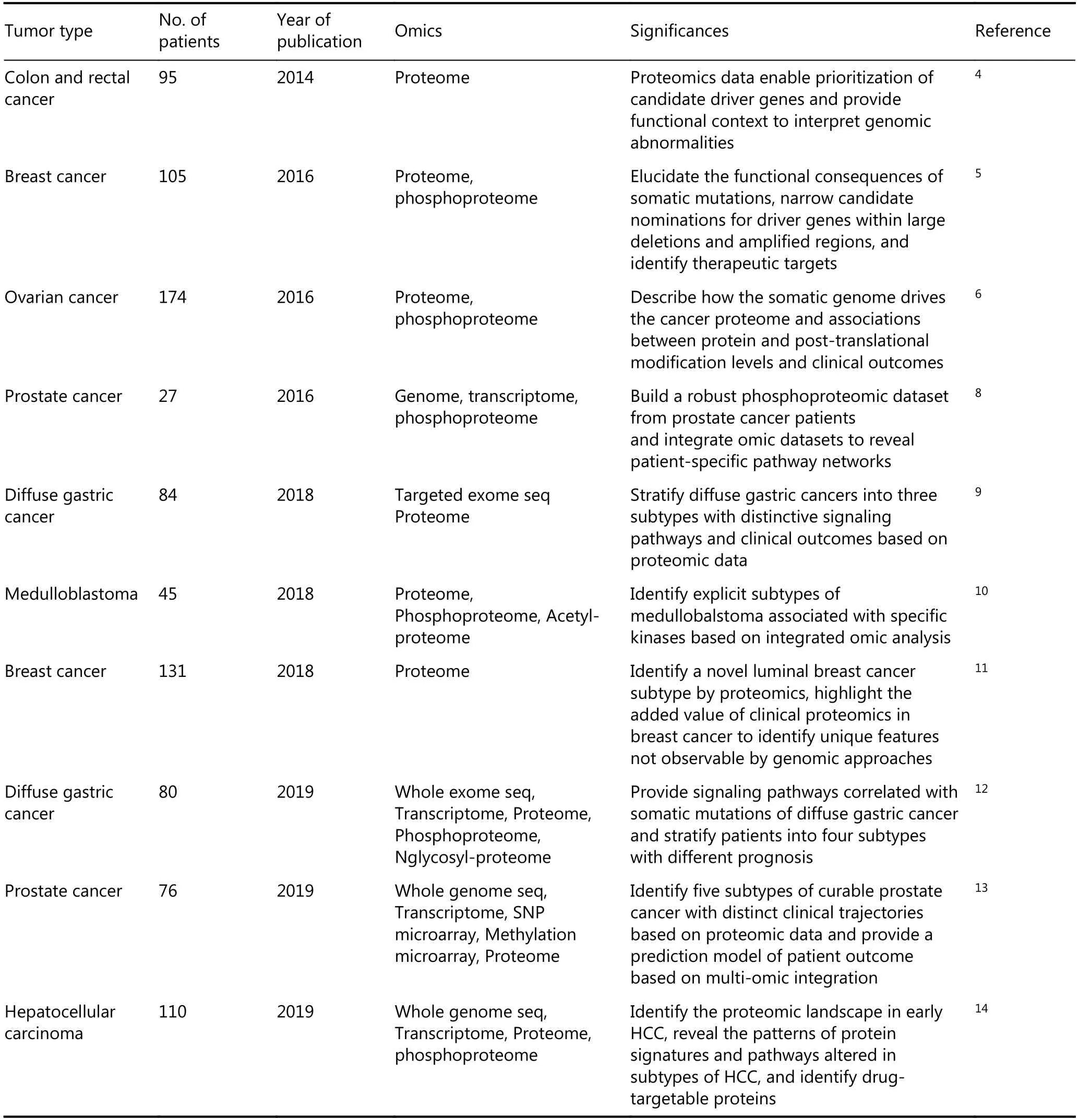
Table 1 Summary of the current reports of large-scale cancer proteomics studies
Proteomic analysis of early-stage hepatocellular carcinoma
Recently, the He group from Beijing Proteome Research Center reported their large-scale proteomic study of earlystage hepatocellular carcinoma14. Hepatocellular carcinoma(HCC) is the most common primary malignancy of the liver,and the main risk factors are chronic hepatitis B and aflatoxin B1 exposure15. An estimated 841,080 new liver cancer cases occurred worldwide in 2018, 46.7% of which occurred in China (Figure 2). Liver transplantation and surgical resection are considered the best approaches to treat HCC. Combined with the use of systemic treatments such as sorafenib and lenvatinib, the survival of patients with HCC has been improved in recent years. However, the prognosis of HCC is still not optimistic, with a five-year overall survival rate of 50%–70%. Therefore, additional information about the molecular characteristics of HCC is urgently needed to develop effective combination therapy strategies.
He and coworkers analyzed 110 paired tumor and nontumor tissues from patients with early-stage HCC related to hepatitis B virus infection using shotgun proteomics. Wholegenome sequencing was performed on 93 paired tissues, and RNA sequencing was performed on 35 paired tissues. Labelfree quantification was conducted with MaxQuant16to describe the qualitative proteome and phosphoproteome of these tissues. Compared to most previous proteogenomic studies that employed iTRAQ or TMT isobaric labeling for protein quantification, the label-free approach is more flexible and less expensive. In addition, tryptic peptides were separated into fewer fractions in this study, which reduced the instrument time.
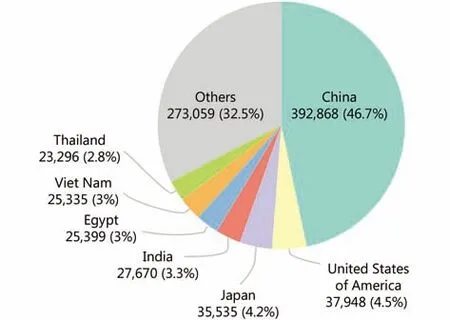
Figure 2 Estimated number of new liver cancer cases in 2018 by International Agency for Research on Cancer (http://gco.iarc.fr/).
First, several carcinogenesis pathways were upregulated in HCC, including the cell cycle. Phosphoproteomics revealed hyper-phosphorylation of proteins involved in inflammation and cell metastasis pathways in these tumors. Additionally,by categorizing the tumors according to the serum αfetoprotein (AFP) level or microscopic vascular invasion(MVI), the authors identified the enrichment of immunityrelated pathways, e.g., TCR, CXCR4, and NFκB, in the AFPhightumors, and proteins involved in the extracellular matrix-receptor interaction and the TGFβ signaling pathways were enriched in MVI+tumors. Interestingly, the in-depth description of differentially expressed proteins in clinically annotated tumors revealed a correlation between protein dysregulation and aberrant genetic events. For example,ribosomal protein S6 (RPS6), a downstream substrate of mTOR, is hyper-phosphorylated in the tumors carrying TSC1/2 loss-of-function mutations. However, this study did not provide a more comprehensive integration of the proteomic data and the sequencing data to determine whether some of the detected somatic mutations were validated at the protein level or the potential correlations between the abundance of mRNAs and the levels of their protein products.
Next, this study clustered the tumors into three subtypes based on protein levels using a non-negative matrix factorization consensus-clustering analysis17, an efficient machine-learning approach for the identification of distinct molecular patterns and molecular classification. Patients in the third cluster, S-III, had a worse prognosis, a higher degree of MVI+, AFPhighstatus, younger age, and significantly lower overall survival rate and higher recurrence rate compared with the other two subtypes. S-I tumors were characterized by increased levels of proteins and pathways related to liver function, and S-II and S-III tumors were both characterized by increased levels of proliferation-related proteins and pathways. Additionally, the S-III tumors were characterized by activated tumor-promotion pathways and higher levels of immune cell infiltration. This study highlights the potential of using proteomic data for molecular subtyping, and the information can be used to predict the prognosis of patients with HCC. For example, patients with the S-III subtype should be provided more aggressive treatments after surgery.More importantly, some of the identified signature proteins may provide additional therapeutic targets for these patients.
Finally, the proteomic data revealed the upregulation of SOAT1, an enzyme that catalyzes the formation of fatty acidcholesterol esters, in HCC tumors, and this change significantly correlated with a poor prognosis. According to the results of a loss of function study, SOAT1 knockdown or treatment with the SOAT1 inhibitor avasimibe suppressed the proliferation and migration of HCC cells by reducing the plasma membrane cholesterol content and inhibiting the integrin and TGFβ signaling pathways. Furthermore, the avasimibe treatment significantly reduced tumor growth in the SOAT1highPDX models, suggesting that avasimibe may be used to treat patients with HCC presenting a high level of SOAT1. Thus, cholesterol homeostasis is dysregulated in HCC, which might be explored as a therapeutic target. This study confirms the value of large-scale proteomic analyses in the discovery of novel therapeutic targets and the potential use of proteomic profiling in personalized medicine.
Taken together, this study utilized MS-based proteomics to depict a comprehensive molecular landscape of early-stage HCC tumors at the protein level and characterized the molecular signatures of three subtypes of HCC by integrating the proteomic data with clinical features. Through these efforts, the authors discovered a subgroup of early-stage HCC with a more aggressive phenotype and the worst prognosis,and identified a number of targetable proteins associated with a poor prognosis. Among these proteins, SOAT1 is a promising treatment target for patients with SOAT1highHCC.This study established a strategy to accelerate drug target discovery using proteomic profiling, underscoring the potential applications of MS-based proteomics in precision medicine for early diagnosis, prognosis predictions,personalized medicine and rational targeted drug selection.
Perspective
All the studies discussed above reported the potential value of in-depth proteomic profiling in the discovery of prognostic biomarkers and druggable targets. Interestingly, proteomic data were recently shown to outperform data describing mutations, CNVs and mRNA expression in identifying the associations between drug and the target proteins or pathways18. By directly analyzing the proteins, researchers may have more opportunities to identify a more responsive and effective cancer treatment strategy. However, the identified signatures from those studies require further evaluation in sufficiently large clinical sample sets before they actually transform the clinical practice of cancer treatment,which requires the global collaborative efforts of proteomics specialists, cancer biologists and oncologists. Additionally,although a large amount of data has been generated in these studies and shared in public databases, better bioinformatics tools are still required to ensure that people without expertise in proteomics are able to easily interpret those data and use the information for their own research.
Compared with NGS based genomics and transcriptomics,MS-based proteomics has its own limitations and challenges,such as the low sequence detection coverage and the inefficient detection of somatic mutations. A typical proteomic strategy employs a single enzyme, trypsin, to digest proteins into peptides by specifically cleaving the proteins after lysine (K) and arginine (R) amino acid residues. Trypsin-digested peptides that are too long or too short may not be observed by the mass spectrometer. In addition, a peptide precursor ion is usually selected for sequencing in MS/MS based on its abundance after elution from a reverse-phase liquid chromatography column.Therefore, less abundant proteins tend to be ignored, leading to a reduced detection coverage. Proteomics has been successfully used to detect some somatic mutations at the peptide level, as well as novel splicing events using genomics and transcriptomics data derived from a patient-specific protein sequence database (the workflow is illustrated in Figure 1). However, only a small fraction of the variants detected at the DNA and RNA levels are observed in proteomic data4,5. One possible explanation is that some gene products containing single amino acid variants (SAAVs),frameshifts, and splice variants are unstable, targeted for degradation, or otherwise untranslated. The detection rate of the existing abnormal peptides is low due to their low abundance and the low detection coverage of the mass spectrometer, as discussed above. The limitations will be improved with advances in instrumentation and further development of sample preparation and data acquisition methods.
The structures, activities and functions of proteins are tightly regulated by their chemical modifications. More than 200 different types of PTMs have been identified in proteins19. The most commonly detected PTMs include phosphorylation, acetylation, glycosylation, and ubiquitination. Accumulating evidence has confirmed the importance of PTMs in the regulation of cancer-related signal transduction ranging from sustaining cell proliferation to cancer metastasis. Another great advantage of MS-based proteomics is the ability to detect PTMs, which are missing in genomics and transcriptomics. Deep coverage of the phosphoproteome was achieved in some of the studies discussed above, and a number of novel phosphosites were reported, improving our understanding of the signaling pathways in various types of cancers. A global phosphosite analysis using MS-based phosphoproteomics enables the reconstruction of cancer-specific or tumor-specific signaling networks, which not only describe the activity of specific kinases but also the crosstalk between different signaling pathways8. This information may be used to design combination therapeutic strategies with optimized efficacy and to identify potential solutions to overcome drug resistance.
However, several challenges remain for the application of MS-based PTM studies. For example, phosphorylation is highly dynamic, and extra caution is required during sample processing and storage to avoid compromising the data. For the same reason, a large number of samples should be evaluated in PTM studies to support robust clinical correlations. Additionally, compared to protein expression profiling, a much larger amount of starting material is usually required for PTM analysis, limiting its clinical applications in biopsies or small tumors. Improved sample preparation strategies and MS analysis methods with higher sensitivity are required to support the clinical application of PTM profiling.Furthermore, currently, the available PTM profiles are still quite limited. Most of these studies report on phosphorylation, but information about other types of PTMs is lacking, preventing an assessment of the clinical value of large-scale PTM profiling.
Clinical proteomics has been integrated with genomics and transcriptomics, and this approach has begun to reveal the comprehensive molecular landscapes of cancer. However,pre-fractionation is usually required before MS analyses to reduce the sample complexity and achieve high coverage of the proteome, which dramatically increases the instrument time and costs, limiting the use of proteomics to analyze large-scale tumor sample sets and its potential applications in clinical practice for personalized medicine. With advances in instrumentation and sample processing methods that may further improve the detection sensitivity and throughput of proteomics, more proteome datasets for different types of cancer are expected to become available in the future,providing crucial information that will improve our understanding of cancer biology. These data will also support different aspects of cancer management, including biomarker discovery, drug target selection, prognosis predictions, etc.
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (Grant No. 21974094,21575103), Tianjin Natural Science Foundation (Grant No.18JCYBJC25200), Young Elite Scientists Sponsorship Program by Tianjin (Grant No. TJSQNTJ-2017-10).
Conflict of interest statement
No potential conflicts of interest are disclosed.
 Cancer Biology & Medicine2019年4期
Cancer Biology & Medicine2019年4期
- Cancer Biology & Medicine的其它文章
- Interpretation of breast cancer screening guideline for Chinese women
- Breast cancer screening guideline for Chinese women
- Erratum to Simultaneous inhibition of PI3Kα and CDK4/6 synergistically suppresses KRAS-mutated non-small cell lung cancer
- The correlation and overlaps between PD-L1 expression and classical genomic aberrations in Chinese lung adenocarcinoma patients: a single center case series
- Nomogram based on albumin-bilirubin grade to predict outcome of the patients with hepatitis C virus-related hepatocellular carcinoma after microwave ablation
- Omics-based integrated analysis identified ATRX as a biomarker associated with glioma diagnosis and prognosis