
The challenge of drug resistance in pancreatic ductal adenocarcinoma: a current overview
2019-02-16 06:07:00FranciscoQuioneroCristinaMesasKevinDoelloLauraCabezaGloriaPerazzoliCristinaJimenezLunaAnaRosaRamaConsolaciMelguizoJosePrados
Cancer Biology & Medicine 2019年4期
Francisco Quiñonero, Cristina Mesas, Kevin Doello, Laura Cabeza, Gloria Perazzoli, Cristina Jimenez-Luna, Ana Rosa Rama, Consolación Melguizo, Jose Prados
1Institute of Biopathology and Regenerative Medicine (IBIMER), Center of Biomedical Research (CIBM), University of Granada, Granada 18100, Spain; 2 Department of Anatomy and Embryology, Faculty of Medicine, University of Granada,Granada 18071, Spain; 3 Instituto Biosanitario de Granada (ibs. GRANADA), Granada 18014, Spain; 4 Medical Oncology Service, Virgen de las Nieves Hospital, Granada 1812, Spain; 5Department of Oncology, Ludwig Institute for Cancer Research,University of Lausanne, Epalinges 1066, Switzerland; 6Department of Health Science, University of Jaén, Jaén 23071, Spain
ABSTRACT Pancreatic ductal adenocarcinoma (PDAC) has one of the highest mortality rates among all cancer types. Its delayed diagnosis precludes curative resection, thus most of the current therapies against PDAC are based on chemo- and radiotherapy.Unfortunately, these strategies are insufficient to improve its poor prognosis. Despite the advances made in chemotherapy (e.g.nab-paclitaxel and gemcitabine), many patients with PDAC are unable to benefit from them due to the rapid development of drug resistance. Currently, more than 165 genes have been found to be implicated in drug resistance of pancreatic tumors, including different integrins, mucins, NF-κB, RAS and CXCR4. Moreover, drug resistance in PDAC is thought to be mediated by the modulation of miRNAs (e.g. miRNA-21, miRNA-145 and miRNA-155), which regulate genes that participate in cell proliferation,invasion and metastasis. Finally, cancer stem cells are intimately related to drug resistance in PDAC due to their ability to overexpress ABC genes -involved in drug transport-, and enzymes such as aldehyde dehydrogenases -implicated in cellular drug metabolism- and poly (ADP-ribose) polymerases -involved in drug-induced DNA damage repair. Understanding the mechanisms involved in drug resistance will contribute to the development of efficient therapeutic strategies and to improve the prognosis of patients with PDAC.
KEYWORDS Pancreatic ductal adenocarcinoma; chemotherapy; drug resistance; cancer stem cells; therapeutic strategies
Introduction
Pancreatic ductal adenocarcinoma (PDAC) has one of the highest mortality rates among all cancer types and increases its incidence year by year. The 5-year survival rate is only 5%,even in patients undergoing complete tumor resection or treated with chemo- and radiotherapy1. PDAC is the most frequent type of pancreatic cancer (PC), affecting 90% of patients with cancer in the pancreas, and it is the third cause of cancer-related death in the United States, following lung and colorectal cancer PDAC has no visible symptoms or biomarkers, which hinders its early diagnosis1-2. As a consequence, more than 50% of patients present with metastatic disease at diagnosis, when no curative treatment can be offered. Many of the currently used drugs may increase the lifetime of patients and relieve their symptoms,but neither cancer eradication nor complete symptomatic relief is usually possible. Few drugs have been shown to be effective against PC over the years. gemcitabine, a nucleoside analogue used since the 90s as the clinical agent of reference2is the most common chemotherapy agent used in clinical practice. Unfortunately, low survival rates are still achieved with this drug. To increase treatment efficiency, formulations of gemcitabine encapsulated in albumin nanoparticles have been assayed in vitro and in vivo3. On the other hand, 5-fluorouracil (5-FU), a molecule widely used in colon cancer treatment due to its capacity to be inserted into the DNA and inhibit cell proliferation, lacks a therapeutic efficacy in PC,where no significant improvement in symptoms or life expectancy was demonstrated4. Clinical studies reporting the use of 5-FU along with gemcitabine did not show clinical benefits in comparison with gemcitabine alone, but a slight increase in side effects (neutropenia, diarrhea and anemia)was reported5. However, FOLFIRINOX, a formulation containing several drugs (including 5-FU), increased the survival of patients with advanced PC by two months6Irinotecan, an inhibitor of Topoisomerase I present in FOLFIRINOX, showed effectiveness in the treatment of this type of cancer in some clinical trials7, and its liposomal encapsulation would improve the treatment of refractory PC8. Finally, Paclitaxel associated with human albumin (nabpaclitaxel) is also being used in PC9. Recently, a clinical trial demonstrated that nab-paclitaxel plus gemcitabine improved the survival of patients with advanced PC by two months,without significantly increasing drug toxicity10. Currently,the most effective therapies in clinical terms are nabpaclitaxel, gemcitabine and folfirinox. However, this regime is not applicable to all patients given the increase in toxicity and the severe risk for patients in advanced stages of the disease.
In most cases, PC progresses to infiltration of other organs and distant metastasis, which have a high impact on survival.In these patients, conventional treatment does not improve the prognosis. A cornerstone of this therapeutic failure is the development of drug resistance. Accumulating evidence suggests that chemoresistance is intimately linked to the disruption of multiple genes involved in intracellular signaling, DNA repair, metabolism and regulation of cell replication11. In addition, local recurrence of the tumor after surgical resection, chemo- and/or radiotherapy has been related to the presence of cancer stem cells (CSCs). These cells are characterized by a high treatment resistance and proliferation capacity, which explains the more aggressive nature of the recurrent tumor12. In this review we analyze different genetic and protein resistance mechanisms by which PDAC cells reduce the efficacy of the available drugs, and the advances being made to avoid such drug resistance and decrease the current mortality rate of PC.
Drug resistance at the molecular level
Although currently gemcitabine is the first-line treatment against PDAC, many patients are unable to benefit from it due to the rapid development of resistance to this drug by the tumor cells11. Gene expression microarray analyses performed in PC cell lines showed more than 165 genes related to drug resistance. These genes were involved in a myriad of cell functions, including antioxidant activity,apoptosis, cell cycle regulation and transduction of signals,among others13(Figure 1). gemcitabine inhibits cell proliferation and induces apoptosis of tumor cells through the activation of the AMPK / mTOR pathway, increasing the expression of AMPK and decreasing that of mTOR, which results in cell autophagy14. In this route, ARK5, a kinase related to AMPK, induces the epithelial-mesenchymal transition (EMT) of PC cells, which is linked to drug resistance. Recently, the inhibition of ARK5 by modulating the oxygen conditions (normoxia/hypoxia) proved to sensitize pancreatic cells to gemcitabine15. The SRC tyrosine kinase, which has been used as a prognostic marker in PC,may also be involved in drug resistance of PC cells16. In fact,some natural compounds that act over this molecule were able to suppress tumor growth and decrease the chemoresistance of tumor cells against gemcitabine17.Similarly, the overexpression of integrin β1, an adhesion molecule involved in the interaction between cells and the extracellular matrix, has been linked to chemotherapy resistance in solid cancers, including PDAC18. This integrin induced gemcitabine resistance by activating CDC42 and the PI3K pathway19, which regulate the cell cycle and apoptosis20.Finally, mutations in the RAS proto-oncogenes -detected in a high proportion of human pancreatic tumors- have been associated with drug resistance21. The RT11-i (an antibody that inhibits the RAS / RAF / MEK and PI3K / AKT signaling pathways) reduced gemcitabine-resistance in PC cell lines22.In addition, the inhibition of RAB14, a member of this family of proteins, decreased the IC50of gemcitabine and increased apoptosis induction23.
On the other hand, two of the most important glycoprotein families, i.e. ABC transporters and mucin proteins, have been related to PDAC resistance. ABC transporters extrude drugs out of the cell, decreasing their intracellular concentration. Cancer cells expressing these transporters are generally referred to as multidrug resistant(MDR) cells24-25. In this vein, overexpression of the ABCB1 gene was found to be deregulated in pancreatic tumors originated due to overexpression of the MYC oncogene26.Similarly, MUC1 overexpression induces the expression of several drug-resistance genes in pancreatic adenocarcinoma27. In addition, this glycoprotein induces the expression of HIF1α and increases the metabolic rate and internalization of glucose. These processes have been implicated in an increased resistance to gemcitabine28.Moreover, the deregulation of MUC4, a member of the MUC family, has been related to the first disturbances that result in carcinogenesis and drug resistance in PC29. In fact, the overexpression of MUC4 was associated with a negative regulation of the expression of a nucleoside transporter(hCNT1) involved in cell internalization of gemcitabine30-31.

Figure 1 Different mechanisms of drug resistance in pancreatic cancer deregulated molecular signal pathways such as RAS, NFκB and PI3K pathways and, overcoat KRAS and BRCA genetic deregulation. These mechanisms comprise that tissular hypoxia increases HIF-1 levels and diminishes reactive oxygen species (ROS), a dense extracellular matrix impedes the diffusion of chemotherapeutic agents, and the existence of cancer stem cells escape to apoptosis.
Some nuclear transcription factors have been implied in drug resistance, as is the case with kappa β (NF-κB), which modulates the immune and inflammatory responses32-33.Similarly, a factor related to NF-κB, named TRIM31, was associated with a more aggressive phenotype of PC in which anti-apoptotic genes involved in gemcitabine-resistance were overexpressed. Therefore, a therapeutic strategy based on specific TRIM31 inhibitors could be useful to decrease gemcitabine-resistance in PC34-35. In addition, gemcitabine can induce the expression of NF-κB and generate reactive oxygen species (ROS) through the activation of the P22 factor. The expression of NF-κB regulates the signaling pathway of CXCR4, another factor that confers resistance against gemcitabine36. The chemokine receptor CXCR4,involved in the first stages of organ development, is of great importance in the tumor genesis and metastatic spread of PC37. Its overexpression is associated with a worse prognosis,probably because the overactivation of the CXCL12-CXCR4 signaling axis confers resistance against current therapies38-39.The basis of this resistance is that CXCR4 negatively regulates the expression of let-7a miRNA, which is responsible for the inhibition of cell proliferation, metastasis and drug resistance39. Another nuclear factor that induces drug resistance is CHK1, a protein able to inhibit the progression of the cell cycle in response to DNA damage. Inhibitors of CHK1 proved to decrease tumor cell resistance and favor the action of antiproliferative drugs (i.e. gemcitabine) in PC cells.Conversely, the inhibition of HSP90 -a CHK1-activating protein- did not increase the sensitivity of PC cells to gemcitabine40. Recently, HSP27, another member of this family, has been demonstrated to be implicated in gemcitabine resistance41-42.
Role of non-coding RNA (ncRNA) in drug resistance
The ENCODE project estimates that ncRNA transcripts constitute approximately 70% of the human genome, having a number of cell regulatory functions. Within ncRNAs,miRNAs regulate 90% of gene expression and influence the processes of cell proliferation, invasion and metastasis.Subsequently, miRNAs have been implied in the diagnosis and prognosis of several cancer types, including PDAC43-45.In addition, some specific miRNAs play a role in the development of drug resistance in PC (Table 1).
For example, miRNA-21 proved to increase drug resistance through the inhibition of FasL expression, a factor that triggers apoptosis. This, in turn, is associated with a decrease in patient survival46. Furthermore, the ability ofmiRNA-21 to induce drug resistance in tumor cells is mediated by the PI3K-AKT pathway, whose activation decreases cell susceptibility to apoptosis through an increased expression of the anti-apoptotic gene Bcl247-48. Likewise,miRNA-29c seems to play a role in chemoresistance of PC cells. Its overexpression is associated with increased levels of USP22, which proved to induce autophagy and inhibition of apoptosis following treatment with gemcitabine49-50.Moreover, the inhibition of miRNA-145 and the increase in miRNA-155 expression have been also related to PDAC drug resistance. The former, a tumor suppressor that increases the sensitivity of tumor cells to gemcitabine, inhibits the signaling pathway of p70S6K1, a protein implicated in drug resistance, tumor growth and metastasis51-52.
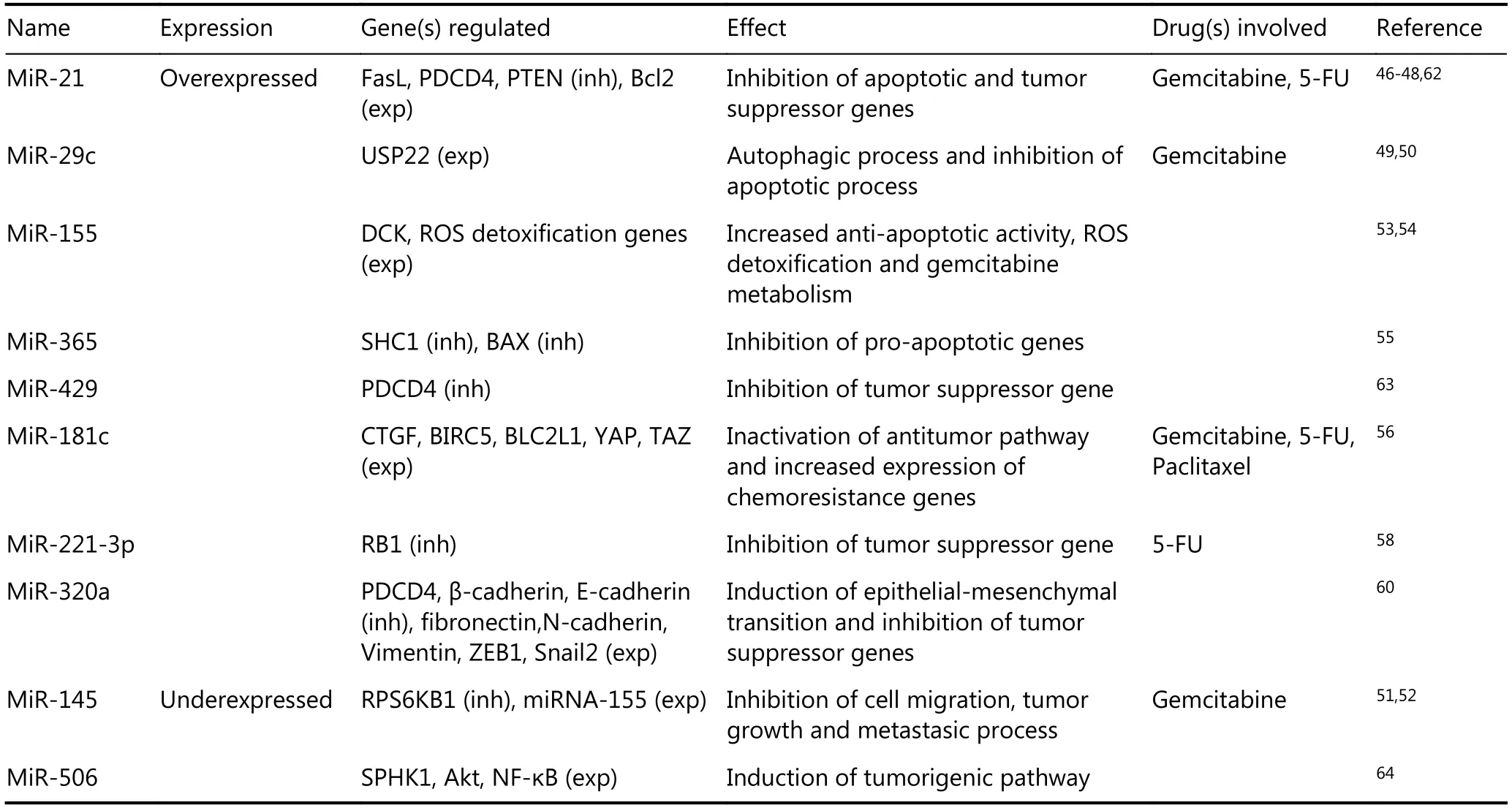
Table 1 miRNAs involved in drug resistance in pancreatic cancer
The latter is involved in the increase in cellular antiapoptotic activity53and in the deregulation of the DCK gene expression (implied in the metabolism of gemcitabine54. In addition, miRNA-155 induces the secretion of exosomes,increases the expression of ROS detoxification genes and decreases DCK expression53-54. Finally, the overexpression of miR-365 through the repression of the pro-apoptotic genes SHC1 and BAX induced gemcitabine-resistance in PC cells55.
Drug resistance in PDAC has been also associated with miRNAs. For example, miRNA-181c, which is highly expressed in advanced stages of PC, increases the chemoresistance against gemcitabine, 5-FU and Paclitaxel by the inactivation of the Hippo antitumor signaling pathway56.Interestingly, the lncRNA GAS5, an ncRNA of more than 200 bp, antagonizes the effect of miRNA-181c and should be explored as a therapeutic strategy57. Moreover, miRNA-221-3p and MiR-320a have been implicated in 5-FU resistance.The former desensitizes PC cells to 5-FU through negative regulation of RB1, a tumor suppressor gene which has been implicated in the development of PC58. The latter inhibits PDCD4, another tumor suppressor gene that increases the expression of molecular markers related to the EMT,promotes the proliferation and migration of tumor cells and makes them more invasive59-60. In fact, PDCD4 is regulated by several miRNAs such as miR18361miRNA-2162and miRNA-42963, which may repress PDCD4 expression in PC cell lines, promoting tumor growth. Finally, the deregulation of miRNA-506 -which acts as a tumor suppressor- boosts the progression of pancreatic tumors, increasing chemoresistance through the modification of the signaling pathway in which NF-κB participates64. In sum, a large number of micro-RNAs are involved in the development of PDAC, acting in certain cases as proto-oncogenes or tumor suppressor genes depending on the signaling pathways regulated.
EMT and drug resistance
Correct adhesion of tumor cells to the cellular matrix is a hallmark of cancer progression. However, many of chemoand radiotherapy-resistant tumors have been proved to originate blood circulating tumor cells from an EMT process65. Cancer stem cells (CSCs) are essential in the genesis of tumors, and they produce a large number of signaling substances involved in cell proliferation and drug resistance. Interestingly, the cells activated during the process of EMT display a gene expression profile similar to that of CSCs, which would explain their ability to form new tumors with great resistance to chemo- and radiotherapy66. In fact,Hangbin et al. were able to sensitize gemcitabine-resistant cell lines through the inhibition of EMT by means of hyperthermia67. One of the metabolic pathways more involved in EMT and drug resistance is PI3K/AKT/mTOR.The deregulation of this pathway causes a decrease in chemotherapy-induced DNA damage, inhibition of apoptosis and a decrease in the expression of E-cadherin –a molecule associated with EMT-. The use of an inhibitor against this signaling pathway allows to inhibit the EMT and the migration of cancer cells, subsequently inhibiting tumor growth, metastasis and EMT in murine models68. Another gene related to EMT is Slug, a transcriptional factor that suppresses E-cadherin expression, which confers resistance to gemcitabine in pancreatic CSCs through EMT. Thus, its suppression at the transcriptional level makes it possible to increase the sensitivity of PC cell lines, reducing their invasive and migratory capacity69. Although different molecular pathways regulate EMT, miRNAs are essential factors in the control of this process. In this vein, miR-509-5p and miR-1243 inhibit the EMT process and their overexpression in PC cells increases the sensitivity to gemcitabine70-71. In conclusion, the use of miRNA inhibitors of EMT, one of the processes that mostly influences drug resistance in PDAC,opens new possibilities in the treatment of this entity.
Role of cancer stem cells in drug resistance
For many years, cancer was thought to be composed of clonal, homogeneous cell populations. Nevertheless, over the years it became evident that tumors are highly heterogeneous systems constituted by cells with varying degrees of differentiation. In fact, it was observed that CSCs, a group of poorly-differentiated cells, are responsible for the selfrenewal capacity of tumors12. The presence of highly drugresistant CSCs in the tumor is a cornerstone in understanding its recurrence (i.e. tumor relapse after chemoor radiotherapy), a phenomenon associated with a worse prognosis72. Although several resistance mechanisms have been described, three systems must be highlighted with regard to PDAC: overexpression of ABC transporters,detoxifying enzymes and proteins involved in cell death processes73.
ABC transporters
The family of ABC transporters is present in most living beings, from the simplest forms of life (bacteria) to the most complex organisms (mammals). These molecules are involved in the transport of different metabolites between the cell membrane and the extracellular matrix against the concentration gradient, using the energy released from the hydrolysis of ATP. Their main functions are detoxification,prevention of intracellular oxidative stress and cell protection against xenobiotics74. However, their detoxification activity serves as an escape mechanism for antitumor drugs and increases resistance to chemotherapy agents (Figure 2).
This resistance is mainly mediated by three receptors:MDR1, BCRP and MRP175. In addition, a high expression of the MRP4 protein has been detected in PDAC. This protein promotes cell proliferation and plays a role in the rapid formation of colonies from tumor cells76. Other genes involved in the synthesis of ABC transporters, such as ABCB4/11, ABCC1/3/5/10 and ABCG2 are also overexpressed in PDAC tissues77. Interestingly, CSCs from PC showed an increased expression of ABC transporters, which is associated with a worse response to chemotherapy. In particular,ABCB1 –which originates a protein known as pglycoprotein- is of major relevance in PDAC as it is considered the ABC transporter more involved in drug resistance, not only in this tumor but in many other cancer types78.
Aldehyde dehydrogenases
The aldehyde dehydrogenases (ALDH) are a family of enzymes whose function is to oxidize cellular aldehydes to carboxylic acids. These aldehydes are originated from the metabolism of several cellular components (proteins, nucleic acids) that often remain as cellular waste, and need to be eliminated. One of the primary functions performed by these enzymes concerns the metabolism of retinol (vitamin A),which is converted into retinoic acid. This molecule is essential for an adequate embryonic development, which makes a high expression of ALDH essential in stem cells79.On the other hand, a great variety of aldehydes are generated from the metabolism of environmental agents and drugs, and they may induce cell damage and death. Therefore, the overexpression of these enzymes protects against these toxic agents and promotes cell survival. In experiments using PDAC cell lines, overexpression of ALDH enzymes allowed to identify cell populations capable of originating tumors more efficiently80.

Figure 2 ABC transporters in detoxification of chemotherapeutic drugs in pancreatic cancer. The most common drugs are oxaliplatin(OXA), 5-fluorouracil (5-FU), gemcitabine (GEM), irinotecan (IRI) and Nab-paclitaxel (Nab-PTX). The main ABC transporters (ATP binding cassette) in pancreatic cancer was breast cancer resistant protein (BCRP), P- glycoprotein (P-gp) and multidrug resistance protein (MRP).MRP and BCRP transporters require the conjugation with glutathione.
Although there are several enzyme isotypes, the ALDH1A1 gene has commonly served as a marker to differentiate normal from CSCs in adult tissues81. Besides, the ALDH1B1 isotype is generally used as a marker of stem cells in the early stages of pancreas development and only a small population of cells that overexpress this marker remains in the adult82.This isotype also promotes tumor proliferation. Accordingly,two phenotypes of PC can be distinguished: those whose growth is favored by the overexpression of ALDH1A1, and those with a dominant ALDH1B1 phenotype83. The inhibition of ALDH1A1 in PDAC cell lines proved to increase sensitivity to gemcitabine, indicating that ALDH1A1 overexpression may be paramount for drug resistance maintenance in tumor cells84. In addition, gemcitabineresistant PDAC cells showed a higher expression of membrane markers also present in CSCs -including ALDH1A1- and an overexpression of the SRC oncogene. The use of an SRC inhibitor along with gemcitabine proved to inhibit tumor proliferation, decreasing the expression of ALDH1A1 and the number of CSCs in the tissue. This indicates that the expression of ALDH1A1 is of significance in both normal and cancer stem cells for the preservation of their phenotype85-86.
The PARP enzyme family
Poly (ADP-ribose) polymerases (PARPs) constitute a family of 18 proteins with a conserved catalytic domain capable of transferring several ADP-ribose units to their target proteins.They are involved in several cellular processes, including the regulation of proliferation and programmed cell death.Moreover, two of the most important members of this family, PARP1 and PARP2, play a role in DNA repair87.Through their catalytic activity, these enzymes modify certain factors responsible for the recruitment of proteins involved in efficient DNA repair (Figure 3). PARP1 is overexpressed in pluripotent cells and its correct expression is essential for maintaining the unique characteristics of human stem cells,including CSCs. Its mechanism of operation is based on the addition of several units of ADP-ribose using NAD+as a substrate, resulting in a poly (ADP-ribose) chain that can contain up to 200 units88. PARP1 modifies p53 and inhibits its binding to the genes that regulate the process of apoptosis.The inhibition of p53 and the repair of DNA damage by PARP may act as mechanisms of drug resistance89.

Figure 3 Mechanism of action of PARP (poly-ADP-ribose-polymerase). Single strand DNA damage induced by chemotherapeutic agents or radiotherapy is repaired by this enzyme leading to cell survival. In this process PARP accumulates ADP tails. However, big amounts of DNA damage cannot be repaired by PARP, so that, ADP tails are released and induce cell death (A). Mechanism of synthetic lethality. BER Base Excision Repair (RES) system repairs single chain DNA damage and Homologous recombination system (HRS) repair double strand DNA damage. These two systems are consecutive, so that, DNA damage and the ineffective of BER lead to the activation of HRS. HRS is made up by repair proteins like BRCA, ATM or ATR. Germinal of somatical mutations in these genes provoke an ineffective HRS dependent DNA repair. This fact could profit to trigger a syntetic lethality, inhibiting PARP with molecules such as olaparib. Therefore, the deffective HRS due to genetic mutations added to BER system inhibition by PARP blockers lead to apoptosis of cancer cells (B).
The overexpression of PARP1 has been associated with different cancer types in humans, including liver, lung,endometrium, ovary and skin90. In 2010, it was discovered that the inhibition of PARP1 diminished cell proliferation in hepatocellular carcinoma by modulating the expression of genes implicated in tumor development and vasculogenesis91.Given the importance of PARP1/2 in DNA damage repair,several drugs aiming to inhibit their activity have been designed as a plausible strategy against cancer. This is the case with olaparib, an inhibitor of both PARP1 and PARP2 that induces S and G2/M arrest and apoptosis. Olaparib has been approved by the FDA since 2014 for the maintenance of patients with ovarian cancer who have mutations in BRCA1 and BRCA292. Simultaneous inhibition of PARP1 and RAD51 proteins, which are capable of interacting with BRCA2 during homologous recombination, has the potential to sensitize cells to radiation therapy, leading to cell cycle arrest and apoptosis93. The use of small molecules that mimic the state of mutated BRCA2 can disrupt the BRCA2-RAD51 interaction, increasing Olaparib effectiveness and allowing the treatment of patients with wild BRCA294. In addition,specific inhibitors against the BET protein family are able to reduce the expression of RAD51, thus increasing sensitization to PARP1/2 inhibitors95. Furthermore, PARP1/2 inhibitors have been used to delay DNA damage repair, allowing sensitization to proton beam irradiation96. Finally, PARP inhibitors have been combined with agents that inhibit telomerase, an enzyme responsible for maintaining telomere length, in order to induce premature aging and apoptosis of PC cells97.
Overcoming chemotherapy resistance
Despite the large number of research lines dedicated to PC,the efficiency of current therapies remains too low. In order to avoid drug resistance, new formulations are being developed based on traditional drugs. One of the main problems with gemcitabine is that its blood concentration is maintained for a short period of time, as the cytidine deaminase breaks it down in just one hour. Therefore, other formulations have been tested to increase drug efficiency. In this vein, the use of PEGylated liposomes allowed to reach a similar tumor concentration of gemcitabine with a 10-fold lower dose, reducing its rapid blood degradation98. Likewise,the use of albumin nanoparticles along with gemcitabine decreased its toxicity and improved its biodistribution and efficiency in in vitro and in vivo assays using PDAC cells99.Other nanoparticles containing gemcitabine and antisense oligonucleotides against the proto-oncogene miR-21 were found to have a high inhibitory effect on the proliferation of PC cells100.
The addition of gemcitabine to cell cultures causes an increase in ROS, leading to apoptosis. However, this process is not totally efficient due to the existence of ROS detoxification enzymes capable of eliminating these molecules. To cope with this mechanism of resistance, Ju et al.101proposed to target the molecular pathways that regulate the expression of detoxification enzymes and use inhibitors against some of these systems (e.g. GSH).Meanwhile, Aibani et al. prevented chemoresistance in PC cells by encapsulating three drugs (5-FU, Leucovorin and Doxorubicin) in PEG particles102. Finally, the use of a plantderived compound (β-sitosterol) together with gemcitabine allowed to efficiently induce apoptosis in pancreatic cell lines through cell cycle arrest in G0/G1 and led to decrease the IC50of gemcitabine, revealing a synergistic effect of both drugs103.
On the other hand, different strategies have been carried out to overcome drug resistance in PC at a clinical stage. For instance, EMT inhibition using antisense oligonucleotides such as Trabedersen has shown positive results in phase I/II clinical trials104. Hyaluronic acid, one of the components of the extracellular matrix, plays an important role in drug resistance in pancreatic adenocarcinoma. Accordingly,hyaluronic acid-degrading enzymes (e.g. hyaluronidase) have been combined with chemotherapeutic agents to improve treatment efficacy, although contradictory outcomes have been reported. In fact, while phase II clinical trials using gemcitabine/Abraxane and hyaluronidase showed significant improvements in terms of progression-free survival, the combination of FOLFIRINOX and hyaluronidase led to poorer overall survival rates105-106.
Finally, although chemotherapy remains as the main treatment in PC, novel immunotherapy-based strategies are showing encouraging results107. Immunotherapy aims to boost the immune response, subsequently increasing tumor cell identification and elimination by the immune system.This can be achieved by means of both passive (e.g.antibodies, activated T-cell transfer) and active techniques(e.g. vaccines)108. However, pancreatic adenocarcinoma has many properties that prevent its recognition by the immune system, including lack of tumor-infiltrating lymphocytes,highly dense extracellular matrix, and production of immunosuppressive cytokines by PC cells. These factors explain why novel immunotherapy treatments (e.g.Ipilimumab, Nivolumab, Pembrolizumab) are not totally effective at present109.
Conclusions
Although remarkable progress has been made in cancer research within the last decade, PDAC still has very low survival rates. The current inability for early detection limits the application of effective treatments. In addition, the development of drug resistance is a key factor to understand the failure of current therapy in both the tumor and metastatic tissues. Drug resistance is mediated by different mechanisms, such as gene mutations involved in critical metabolic pathways and ncRNAs that modulate the expression of genes implied in cell behavior. On the other hand, CSCs from PDAC show a high drug resistance owing to several reasons, including overexpression of PARP enzymes, ABC transporters involved in drug elimination from the cell, and intracellular detoxification enzymes such as ALDHs. Therefore, the increase in survival of patients with PDAC should occur not just by means of discovering early serum markers, but rather due to the development of therapeutic strategies aimed to eliminate pancreatic CSCs and minimize drug resistance.
Acknowledgements
This work was funded by grants from Instituto de Salud Carlos III (Grant No. DTS15/00201 and DTS17/00081) and Junta de Andalucía (Grant No. PIN-0474-2016).
Conflict of interest statement
No potential conflicts of interest are disclosed
 Cancer Biology & Medicine2019年4期
Cancer Biology & Medicine2019年4期
- Cancer Biology & Medicine的其它文章
- Interpretation of breast cancer screening guideline for Chinese women
- Breast cancer screening guideline for Chinese women
- Erratum to Simultaneous inhibition of PI3Kα and CDK4/6 synergistically suppresses KRAS-mutated non-small cell lung cancer
- The correlation and overlaps between PD-L1 expression and classical genomic aberrations in Chinese lung adenocarcinoma patients: a single center case series
- Nomogram based on albumin-bilirubin grade to predict outcome of the patients with hepatitis C virus-related hepatocellular carcinoma after microwave ablation
- Omics-based integrated analysis identified ATRX as a biomarker associated with glioma diagnosis and prognosis