
峨眉山与黄山藏酋猴肠道菌群组成的比较
2019-02-15 05:09翟子豪宋飏王俊茵张可俊孙丙华李静
四川动物 2019年1期
翟子豪, 宋飏, 王俊茵, 张可俊, 孙丙华, 李静*
(1. 四川大学生命科学学院,生物资源与生态环境教育部重点实验室,四川省濒危野生动物保护生物学重点实验室,成都610065; 2. 峨眉山景区管委会,峨眉山生物多样性保护研究所,四川峨眉山614200;3.安徽大学资源与环境工程学院,安徽省黄山生物多样性与短尾猴行为生态学国际联合研究中心,合肥230039)
哺乳动物通过母体产道出生时,首次获得肠道微生物(Vaishampayanetal.,2010)。随着时间的推移,在食物、社会接触或周围环境的影响下,肠道微生物多样性逐渐增加(Davidetal.,2014)。这些微生物之间及微生物与宿主之间形成了相互依存、相互作用的不可分割的整体。微生物菌群和宿主相互交换能量物质、传递信息,在宿主的营养、免疫、代谢中起重要作用(Egertetal.,2006)。随着食物、生境等的改变,肠道微生物的组成也会发生相应的变化。研究表明,在不同季节(Sunetal.,2016)、圈养与野生(Wangetal.,2016)、不同海拔(Zhaoetal.,2018)的条件下,同种动物的肠道微生物组成均有明显的差异。Clayton等(2016)发现,野生白臀叶猴Pygathrixnemaeus和鬃毛吼猴Alouattapalliata具有明显不同的肠道菌群,而在人工圈养条件下却表现出了较大的相似性,在人体中占主导地位的普氏菌属Prevotella和拟杆菌属Bacteroides物种均增加。Zhao等(2018)发现高海拔地区的恒河猴Macacamulatta较低海拔地区的肠道微生物多样性更高,且有多种独有的可操作分类单元(operational taxonomic units,OTUs)。
藏酋猴Macacathibetana,又称毛面短尾猴、大青猴,隶属于灵长目Primates猴科Cercopithecidae猕猴属Macaca,是我国特有的灵长类,国家Ⅱ级重点保护野生动物,世界自然保护联盟(IUCN)濒危物种红色名录近危(NT)物种(Long & Richardson,2008),《中国脊椎动物红色名录》易危(VU)物种(蒋志刚等,2016)。由于适宜栖息地不断缩小,藏酋猴现主要分布在四川、安徽、贵州、福建等地,分为4个亚种:指名亚种M.t.thibetana、贵州亚种M.t.guizhouensis、黄山亚种M.t.huangshanensis和福建亚种M.t.pullus(蒋学龙等,1996)。
峨眉山和黄山分别位于我国西南部和中东部地区,不同的地理位置塑造了两地独有的自然环境。黄山植被主要为次生常绿阔叶林,峨眉山植被以亚热带常绿阔叶林为主。同时,两地都以其得天独厚的藏酋猴资源开展了灵长类生态旅游,并且两地管理部门都对游客向藏酋猴的投喂行为做出了限制(黄山不允许游客投喂,由管理人员定时、定点、定量投食;峨眉山允许游客购买特定的猴粮投喂)。目前,关于黄山、峨眉山藏酋猴的研究主要集中在栖息活动范围(Zhao,1994)、形态和社群行为(Zhao,1997)等方面,发现两地藏酋猴的猴群大小差异较大(孙丙华等,2010);Sun等(2016)报道了不同季节的黄山藏酋猴肠道微生物在组成和多样性上均有显著差异。但两地猴群肠道微生物组成上的差异迄今尚无相关报道。
本文以16SrRNA基因的V3-V4高变区为分子标记,使用高通量测序技术研究了峨眉山藏酋猴群体的肠道微生物,并与黄山藏酋猴的数据进行比较分析,为揭示藏酋猴肠道微生物的特征提供资料,也为藏酋猴的保护与管理提供科学依据。
1 材料与方法
1.1 研究地点和样本信息
峨眉山位于四川省峨眉山市境内,地理位置103°10′~103°37′E,29°16′~29°43′N,最高海拔3 099 m;黄山位于安徽省南部黄山市境内,地理位置118°01′~118°17′E,30°01′~30°18′N,最高海拔1 864.8 m,两地相距超1 460 km。峨眉山藏酋猴(EM)31个粪便样本采集于生态猴区和雷洞坪,采样时间为5月。在猴群每天出现的游步道守候,等待猴群进食完毕,采集新鲜粪便,回到住所后立即置于液氮中保存。为确保所有样本均来自不同个体,样本间距需大于1.5 m。此外,通过本实验室筛选的微卫星位点进一步确认,共有25只不同个体的样品。黄山藏酋猴(HS)样本下载于NCBI(GenBank 登录号:SRP073002),共25个样品,为黄山野生猴谷的鱼鳞坑YA1群体,采样时间为春季,测序平台为Illumina MiSeq PE250,共产生290 542条高质量序列,平均长度为415.28 bp±9.81 bp(Sunetal.,2016)。
1.2 DNA提取、PCR扩增
取粪便内部部分,使用MoBio PowerSoil®DNA Isolation Kit(12888)提取DNA。16SrRNAV3-V4区的PCR扩增引物为338F(5’-ACTCCTACGGGAGGCAGCA-3’)和806R(5’-GGACTACHVGGGTWTC-TAAT-3’)。反应体系25 μL:Q5 high-fidelity DNA polymerase 0.25 μL,5×Reaction Buffer 5 μL,5×High GC Buffer 5 μL,dNTP(1×104μmol)0.5 μL,模板DNA 1 μL,正、反向引物(10 μmol)各1 μL, ddH2O 11.25 μL。PCR程序:98 ℃ 2 min;98 ℃ 15 s,55 ℃ 30 s,72 ℃ 30 s,27个循环;72 ℃ 5 min。扩增产物使用2%琼脂糖凝胶电泳检测,使用Axygen凝胶回收试剂盒回收目的片段。采用Illumina MiSeq PE250平台对粪便微生物群落DNA片段进行双向测序,原始测序数据与下载的黄山数据合并后进行后续分析。
1.3 数据处理
使用Perl语言编写脚本,采用滑动窗口法对FASTQ格式的双端序列作质量控制:窗口大小为10 bp,步长为1 bp,从5’端第一个碱基位置开始移动,要求窗口中碱基平均质量≥Q20,从第一个平均质量值低于Q20的窗口处截断序列,且截断后的序列长度≥150 bp,不允许存在模糊碱基N。使用FLASH(Magoc & Salzberg,2011)对通过质量初筛的双端序列进行拼接,之后根据每个样本所对应的Barcode序列,使用QIIME 1.9.1(Caporasoetal.,2010)将拼接后的序列按样本分开。扩增子测序结果中序列重复度高,并且大量出现1次或几次的序列,统计学和功能上意义不大。因此,使用Usearch(Edgar,2010)去冗余并过滤低丰度序列,得到非冗余序列后,调用silva.gold数据库(Quastetal.,2013)去除嵌合体序列,之后选用Uparse算法(Edgar,2013)按97%的序列相似度进行聚类和OTUs划分,并生成代表性序列。随后,与Greengenes数据库(Release 13.8)(McDonaldetal.,2012)比对,获取每个OTU所对应的分类学信息,并生成具有注释信息的OTUs表。
1.4 统计分析
去除丰度值低于全体样本测序总量0.01%的OTUs(Bokulichetal.,2013),通过mothur(Schlossetal.,2009)绘制稀疏曲线。使用Clustal Omega(Sieversetal.,2011)对代表序列进行多序列比对,数据过滤后,使用FastTree 2.1.9(Priceetal.,2009)构建系统发育树。对OTUs进行拉平处理,之后计算每个样本的α多样性和β多样性。α多样性由反映群落丰富度的ACE指数、Chao1指数,反映群落多样性的Simpson指数、Shannon指数和反映样品覆盖度的Good’s coverage指数进行评估,并通过箱线图进行可视化。β多样性选择Unweighted UniFrac距离(Lozuponeetal.,2011)对样本进行比较,通过主坐标分析(principal co-ordinates analysis,PCoA)、主成分分析(principal component analysis,PCA)和样本的UPGMA聚类分析进行可视化表达。使用LEfSe(Segataetal.,2011)进行组间丰度差异的可视化,首先采用非参数因子Kruskal-Wallis秩和检验检测组间丰度差异显著的物种,之后对这些物种进行成对Wilcoxon秩和检验,最后通过线性判别分析(LDA)对数据进行降维和评估差异显著的物种的影响力。采用PICRUSt(Langilleetal.,2013)对序列进行基因功能预测分析,并使用LEfSe进行组间差异可视化。本研究中的显著性水平检验均选用单因素方差分析(One-Way ANOVA,α=0.05)。
2 结果
2.1 测序数据
经质量控制后,50个样品共得到高质量序列714 259条[(14 285条±3 274条)/样品],其中,EM为 423 717条,HS为290 542条,每条序列平均长度为413.87 bp±12.34 bp。序列按97%相似度聚类后得到500个OTUs,其中,EM独有119个,HS独有34个,两地共有347个(分别占峨眉山和黄山的74%和91%)。按样品最低序列数(4 203条)进行拉平处理。Good’s coverage指数平均值为99.15%±0.14%(98.76%~99.46%),表明99%的样品均得到了鉴定。50个样品的稀疏曲线趋于平缓(图1),表明测序数据量已覆盖样本中的绝大多数物种。
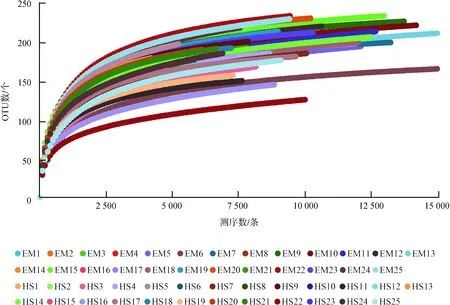
图1 峨眉山藏酋猴和黄山藏酋猴各样品稀疏曲线
Fig. 1 The rarefaction curves of each sample ofMacacathibetanabetween Mount Emei and Mount Huangshan
2.2 EM与HS肠道微生物组成
Greengenes数据库(Release 13.8)匹配到的OTUs可划分为9门46科79属。其中,EM肠道中,相对丰度大于1%的门有8个:厚壁菌门Firmicutes(69.04%±11.81%)、拟杆菌门Bacteroidetes(21.59%±10.05%)、放线菌门Actinobacteria(2.73%±2.17%)、螺旋体门Spirochaetes(1.69%±2.74%)、蓝藻菌门Cyanobacteria(1.49%±2.60%)、变形菌门Proteobacteria(1.27%±1.75%)、软壁菌门Tenericutes(1.16%±1.75%)、疣微菌门Verrucomicrobia(1.00%±1.72%),占门水平总丰度的99.97%;HS肠道中,相对丰度大于1%的门有3个:厚壁菌门(46.34%±8.15%)、拟杆菌门(36.75%±6.38%)、变形菌门(14.91%±8.06%),占门水平总丰度的97.97%。EM肠道中,厚壁菌门与拟杆菌门相对丰度的比值(3.20)显著高于HS(1.26,P<0.05)。在属级水平上,EM肠道中相对丰度最高的为颤螺菌属Oscillospira(23.49%±16.63%),其次为普氏菌属(19.94%±14.48%)、柔嫩梭菌属Faecalibacterium(18.40%±12.41%);HS肠道中相对丰度最高的为普氏菌属(36.35%±9.15%),其次为琥珀酸弧菌属Succinivibrio(13.94%±11.47%)、颤螺菌属(13.76%±10.59%)(图2)。
为了比较两地藏酋猴肠道微生物的差异,选用LEfSe发掘在丰度上有显著差异的类群。在EM中,厚壁菌门、梭菌纲Clostridia、梭菌目Clostrisiales、瘤胃菌科Ruminococcaceae和毛螺菌科Lachnospiraceae均显著富集,主要表现在布劳特氏菌属Blautia、粪球菌属Coprococcus、颤螺菌属、瘤胃球菌属Ruminococcus等的增加,同时螺旋体门的螺旋体纲Spirochaetia、螺旋体目Spirochaetales、螺旋体科Spirochaetaceae、密螺旋体属Treponema也在EM中显著增加(|LDA score|>4且P<0.05)(图3)。而在HS中,拟杆菌门的拟杆菌纲Bacteroidia、拟杆菌目Bacteroidales、普雷沃氏菌科Prevotellaceae、普氏菌属,变形菌门的胃螺杆菌属Flexispira和琥珀酸弧菌属均显著富集。
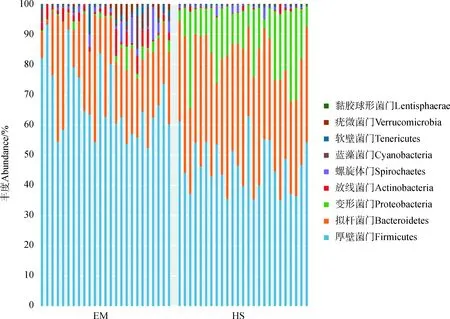
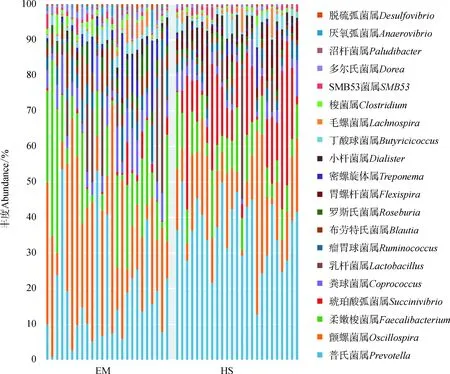
图2 峨眉山与黄山藏酋猴肠道菌群中门(上)和属(下)的相对丰度Fig. 2 Relative abundance of gut microbiome taxa on phylum (upper) and genus (bottom) category in Macaca thibetana from Mount Emei and Mount Huangshan
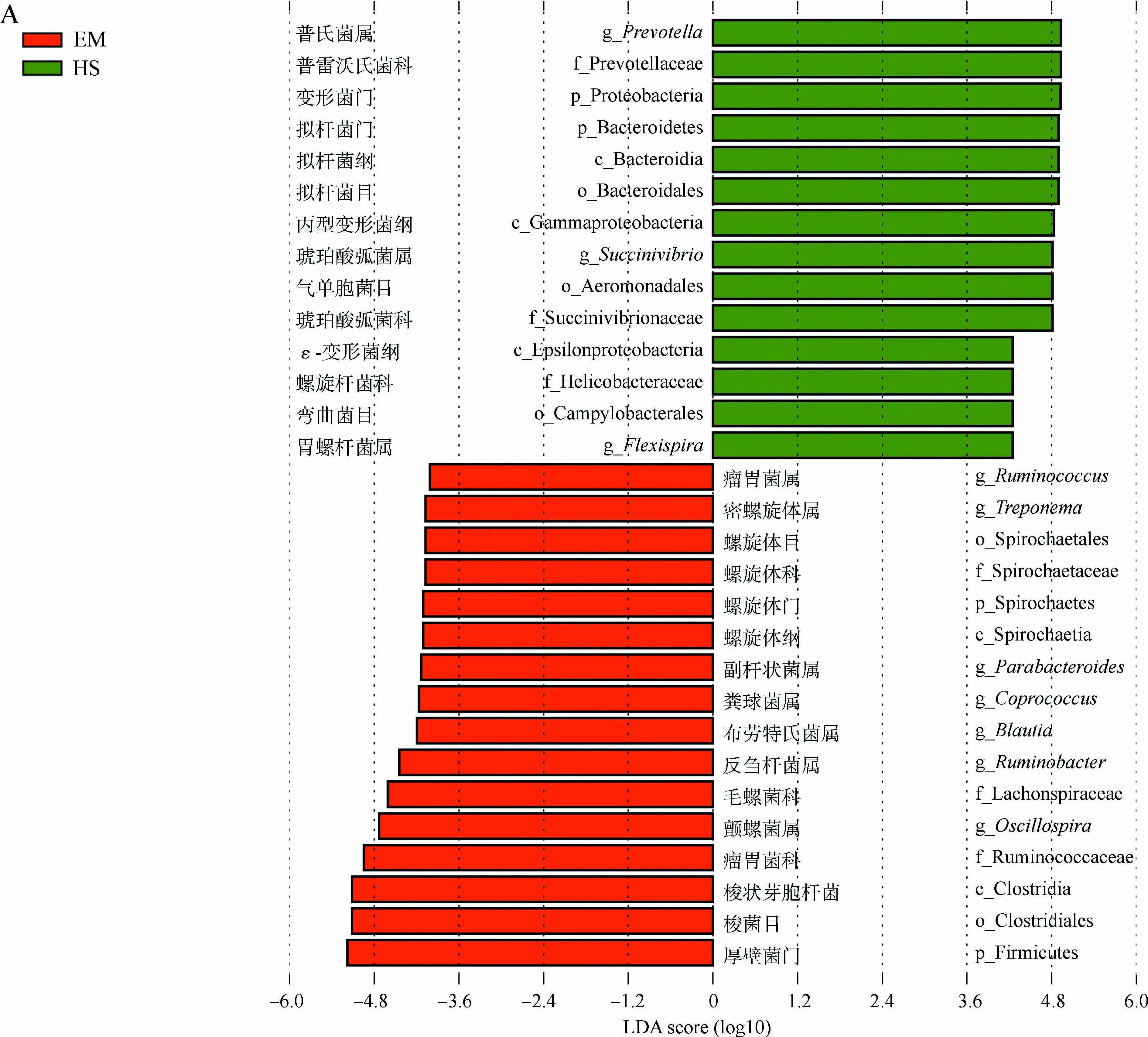
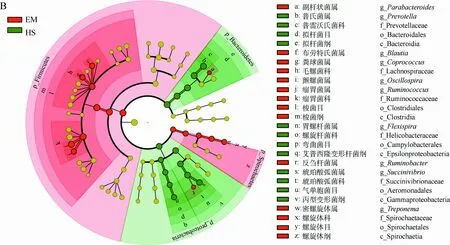
图3 LEfSe分析峨眉山和黄山藏酋猴肠道微生物组成的差异Fig. 3 Analysis of gut microbial composition in Macaca thibetana from Mount Emei and Mount Huangshan based on LEfSe
A. LDA值分布柱状图, B. 进化分支图: 由内到外辐射的圆圈代表由门至属的分类级别, 圆圈的大小代表相对丰度的大小, 节点代表起重要作用的微生物类群, 黄色代表差异无统计学意义的类群
A. LDA value distribution histogram, B. evolutionary branching diagram: the circles from the inside to the outside represent the classification level from the phylum to the genus, the size of the circle represents the relative abundance level, the node represents the important microbial group, and the yellow node represents microbiomes of no significant differences
2.3 EM与HS肠道微生物多样性
EM的ACE指数(P<0.05)、Shannon指数(P<0.01)均显著高于HS,而Chao1指数(P>0.05)和Simpson指数(P>0.1)虽然在EM中较高,但在组间的差异无统计学意义(图4)。PCA、PCoA分析结果均表明,EM与HS在第一轴均明显分开(图5)。
2.4 EM与HS肠道微生物基因功能分析
PICRUSt对EM和HS微生物群落在KEGG代谢途径的差异分析结果显示,共匹配到41个KEGG二级代谢通路,其中差异有统计学意义的代谢通路15个(|LDA score|>2,P<0.05)。EM显著富集的通路5个:脂代谢、外源化学物的生物降解与代谢、信号转导、转录、膜运输;HS显著富集的通路10个:多糖生物合成与代谢、核苷酸代谢、辅因子及维生素代谢、能量代谢、细胞通路和信号、细胞生长与死亡、萜类化合物及多肽代谢、氨基酸代谢、运输和分解代谢、信号分子与互作。
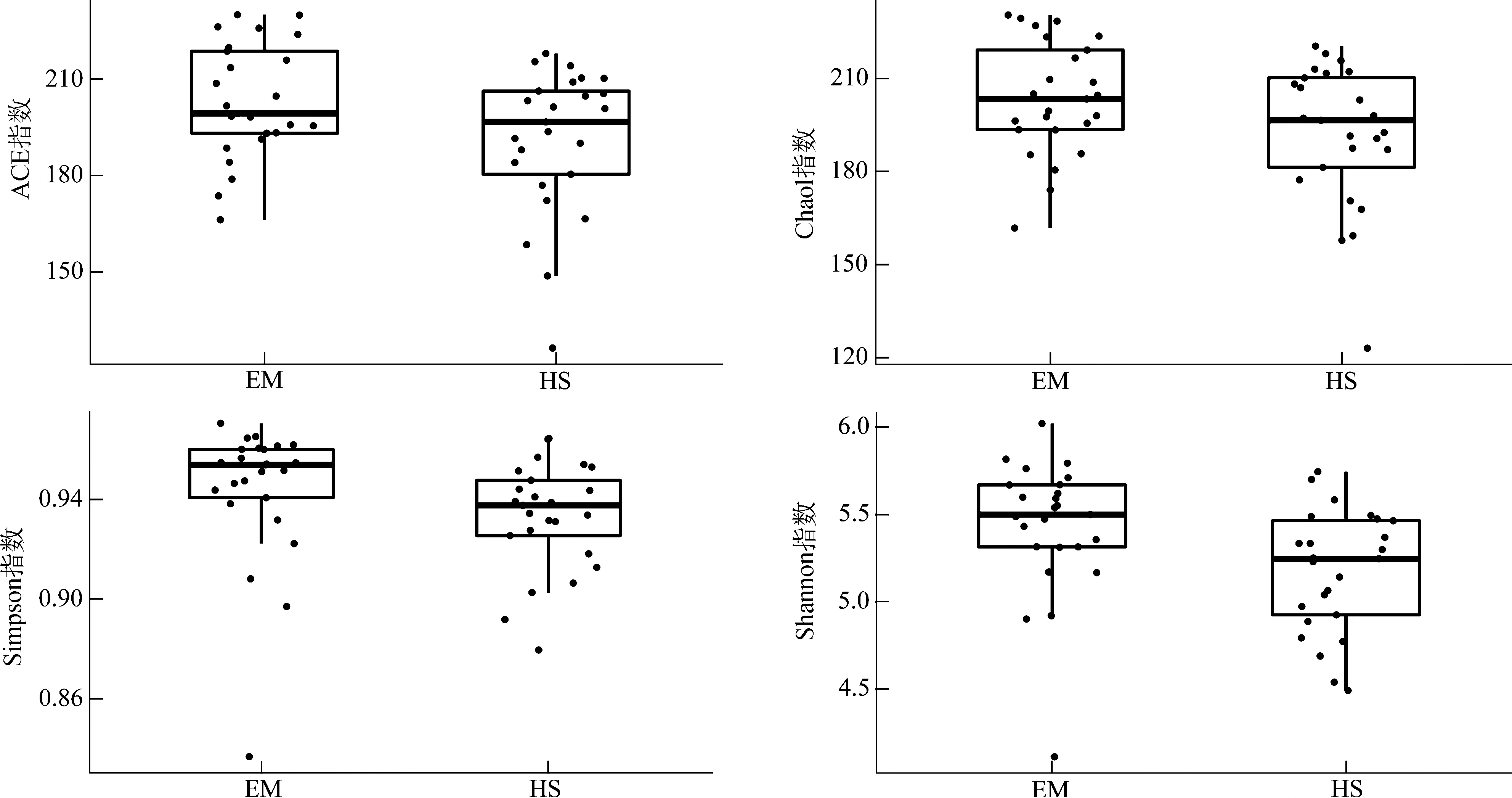
图4 峨眉山藏酋猴和黄山藏酋猴肠道微生物α多样性指数Fig. 4 α diversity index of gut microbiome in Macaca thibetana from Mount Emei and Mount Huangshan
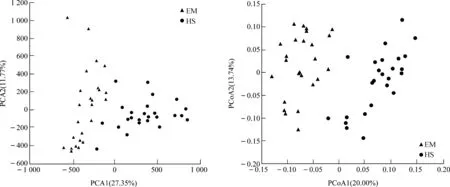
图5 基于PCA和PCoA分析峨眉山和黄山藏酋猴肠道菌群的差异Fig. 5 Difference in the gut microbiome of Macaca thibetana between Mount Emei and Mount Huangshan based on PCA and PCoA
3 讨论
宿主的生活环境、饮食组成决定了其肠道微生物的组成,而肠道微生物组成的变化也反过来影响宿主的健康、食物消化、营养获取。对EM和HS肠道菌群的比较研究有利于了解动物对不同生境的适应性进化,也有利于评估不同生态旅游管理模式对藏酋猴的潜在影响。本研究发现,EM和HS的肠道微生物组成以厚壁菌门、拟杆菌门、变形菌门、放线菌门为主,符合脊椎动物肠道微生物组成的共同特征(Deng & Swanson,2015),也与其他非人灵长类动物相似,如恒河猴(Yasudaetal.,2015)。EM和HS共有的OTUs数量分别占其总数量的74%和91%,表明藏酋猴肠道微生物群落结构相对稳定。
EM和HS肠道微生物在组成、丰度和多样性方面存在明显的差异。峨眉山和黄山地理位置的差异造就了两地不同的植被分布,而这也影响着藏酋猴食物的组成。藏酋猴食性复杂,通常以枝叶、果实、树根等为主,也取食某些昆虫(Bermanetal.,2007)。尤硕愚(2013)报道HS取食共计26科50种植物,主要包括禾本科Poaceae、壳斗科Fagaceae、樟科Lauraceae、杜鹃花科Ericaceae、金缕梅科Hamamelidaceae、山茶科Theaceae、豆科Leguminosae等,其中以壳斗科、樟科的取食量尤为突出,占全年总取食量的51.26%~59.75%。春季时,它们还偏爱取食竹笋,在食物中占据一定比例(熊成培,1984)。而EM取食的植物则高达177种,主要包括五加科Araliaceae、壳斗科、禾本科、木通科Lardizabalaceae、荨麻科Urticaceae等,此外,EM还被观察到取食地面小型无脊椎动物(Zhaoetal.,1991)。食物组成上的差异可能是两地藏酋猴肠道菌群组成和多样性产生差异的主要原因。无论是评价丰富度的ACE指数还是评价多样性的Shannon指数,EM均显著高于HS,这可能与峨眉山丰富的食物种类密切相关。而HS肠道中具有较高丰度的普氏菌属,它们在分解利用水果、谷类和嫩叶中的半纤维素、果胶、淀粉、单糖等时发挥着重要作用(Russell & Baldwin,1979),这可能与HS摄食更多的谷物、竹笋等有关。而EM肠道中富集的颤螺菌属和瘤胃球菌属则可能与食物中的高比例纤维含量相关(Jindouetal.,2006)。
峨眉山和黄山在藏酋猴生态旅游管理方式上的显著不同也是导致两地藏酋猴肠道微生物组成差异的重要原因之一。黄山为吸引藏酋猴到固定地方供游客观赏,管理人员会每天定点定量喂食6 kg玉米(Mathesonetal.,2006),人工食物已成为藏酋猴的固定食物组成;峨眉山则允许游客购买指定的猴粮(主要为花生和红薯干)自行投喂,因此,藏酋猴获取人工食物的量会随着季节和人流量而波动,一般夏秋季居多,冬季最少(本实验调查)。长期固定的人工饮食模式可能导致动物肠道菌群α多样性降低(Uenishietal.,2007),这可能也是HS肠道菌群多样性较EM低的原因。此外,EM具有较高丰度的厚壁菌门与较低丰度的拟杆菌门。厚壁菌门微生物携带许多编码能量代谢相关酶的基因,并且可以产生许多消化酶来分解各种物质,从而帮助宿主消化和吸收养分(Nuriel-Ohayonetal.,2016)。拟杆菌门则在蛋白质、碳水化合物(特别是多糖)等其他能量物质吸收方面发挥着重要作用,从而增加宿主中养分的利用率(Backhedetal.,2004)。研究表明,在经常食用高脂饮食的西方人中,会出现厚壁菌门丰度增加、拟杆菌门丰度减少的现象(Filippoetal.,2010)。而高厚壁菌门、低拟杆菌门也是肥胖人群肠道菌群的重要表现(Clarkeetal.,2012)。肠道菌群KEGG功能分析也发现,EM在与脂类代谢有关的功能通路上有更多的富集,这可能是由于峨眉山游客投喂的猴粮为花生等脂类含量较高的食物,脂类摄入量的增加导致EM肠道中厚壁菌门类群增多,并加强了其对脂类食物的代谢。此外,野外观察也发现部分体型较胖的个体,但这是否与游客投喂食物有关尚需进一步研究。
值得注意的是,在EM中发现了一定丰度的螺旋体门和密螺旋体属,其通常被认为是潜在的病原体(Schwan,1996),可能导致各种慢性传染性皮肤病,如雅司和品他病(Karlssonetal.,2013)。作为灵长类生态旅游的重要组成部分,近距离观赏藏酋猴越来越受到追捧。在管理上,黄山实施人猴隔离,禁止人猴接触及游客投喂;而EM则是逗留在游步道旁,与游客直接接触,给疾病传播提供了条件,可能给灵长类动物和游客都带来潜在威胁(Pedersen & Davies,2009)。KEGG功能分析也发现,EM肠道菌群在外源化学物的代谢与降解途径有显著的功能富集。外源化学物主要包括一些人工合成物或由外界环境摄入,而非机体代谢产生的内源物质。而游客与藏酋猴的直接接触或投喂不适当的食物都可能是导致它们在该代谢通路活动增强的原因。
通过比较和分析峨眉山和黄山藏酋猴的肠道菌群组成,发现两地的藏酋猴在厚壁菌门、拟杆菌门等肠道微生物组成、丰度,以及多样性上存在显著差异,这可能与两地藏酋猴在食物组成、生态旅游的管理模式上的差异密切相关。本研究为藏酋猴的保护和合理开发生态旅游提供了科学参考。
致谢:感谢峨眉山景区管委会郝国歉在采样过程中给予的支持与帮助,谨此致谢。
猜你喜欢
红蜻蜓·中年级(2022年11期)2022-11-23
汽车实用技术(2022年11期)2022-06-20
中国农学通报(2022年14期)2022-06-01
油气田环境保护(2022年2期)2022-05-09
世界竹藤通讯(2020年5期)2020-11-11
作文评点报·低幼版(2020年36期)2020-09-17
文苑(2020年5期)2020-06-16
中国沼气(2019年1期)2019-04-13
世界家苑(2018年8期)2018-09-04
快乐作文·低年级(2017年2期)2017-03-29
