
响应面法制备格列美脲环糊精包合物
2019-01-30 06:49:18赵科郭兆元罗敏徐文霞刘源任静
中国抗生素杂志 2019年1期
赵科 郭兆元 罗敏 徐文霞 刘源 任静,*
(1 抗生素研究与再评价四川省重点实验室,四川抗菌素工业研究所,成都大学,成都 610052;2 成都理工大学,成都 610059)
格列美脲(glimepiride, GM)是近年开发的临床评价最优的磺脲类药物,具有高效、长效、治疗剂量少、不良反应小等诸多优点,临床上应用广泛除了降血糖的作用外[1-2],有研究报导其有可以抑制人乳腺癌细胞增殖的作用[3]。但是格列美脲溶解性低,影响了其在体内的口服吸收;而包合材料β-环糊精(β-CD)的衍生物羟丙基-β-环糊精(HP-β-CD)热稳定性强,水溶性好,对肾脏无毒性,对黏膜和肌肉几乎没有刺激性,溶血作用低,是目前最有前景的药物载体材料之一[4]。因此,可以采用制剂手段,制备格列美脲包合物以提高药物的溶解性并改善其生物利用度[5]。
为了易于操作便于工业化生产,本文采用GM与HP-β-CD通过研磨的方式进行包合,使两者紧紧包合在一起,并通过Box-Behnken响应面法(Box-Behnken design response surface methodology, BBD-RSM)优化格列美脲HP-β-CD片的处方,以期得到最佳处方,且能为格列美脲片的进一步开发奠定良好的理论基础。
1 材料与方法
1.1 试药与仪器
GM对照品(批号:100674-201102,中国食品药品检定研究院),GM原料药(批号:140132-319334,武汉远成共创科技有限公司);乳糖Flow-100(Lactose Flow-100,德国Meggle公司),微粉硅胶和羧甲基淀粉钠(湖州展望药业有限公司),HP-β-环糊精(武汉诺梵生物科技有限公司),微晶纤维素、聚维酮K30、硬脂酸镁(安徽山河药用辅料股份有限公司),羟丙基甲基纤维素(上海卡乐康包衣技术有限公司),其他辅料均为药用级;甲醇、乙腈为色谱纯,其他化学试剂均为分析纯。
LC-10A高效液相色谱仪(含LC-10AT泵和SPD-10A检测器,日本岛津公司);2000傅立叶红外波谱仪(美国PerkinElmer公司);BS224S型电子天平(北京赛多利斯科学仪器有限公司);SHZ-22水浴恒温振荡器(江苏省太仓县医疗器械厂);TDP-5T型单冲压片机(上海超亿制药机械设备有限公司);CJY-2C型片剂脆碎硬度测定仪(上海黄海药检仪器有限公司);RC806D溶出试验仪(天津市天大天发科技有限公司)。
1.2 方法学验证
1.2.1 色谱条件
通过查阅GM有关文献[6-7]及预实验确定其色谱条件:Hypersil BDS-C18柱(200mm×4.6mm, 5μm),流动相为乙腈:0.1%磷酸二氢钠(60:40),检测波长为228nm,流速为1.0mL/min,进样量为20μL,柱温为室温。
1.2.2 溶液的配制
对照品溶液:精密称取GM对照品置于容量瓶中,加适量80%乙腈溶液,超声溶解后,放冷,用80%乙腈稀释定容至刻度,摇匀,即得400μg/mL GM对照品贮备液。
供试品溶液:精密称取GM包合物片剂研磨粉置于容量瓶中,加适量80%乙腈溶液,超声溶解后,放冷,用80%乙腈稀释定容至刻度,摇匀,用0.45μm滤膜进行过滤,即得20μg/mL的供试品溶液。
1.2.3 标准曲线及线性范围
精密吸取适量对照品贮备液,配制成0.1、0.5、1.0、5.0、10、20、40和80μg/mL对照品溶液,进样测定,在波长228nm处用高效液相色谱仪测定其峰面积,以峰面积为纵坐标,浓度为横坐标进行线性回归处理。
1.2.4 精密度
取浓度为20μg/mL的GM对照品溶液,在相同条件下,连续进样6次,测定其峰面积,考察方法的精密度。
1.2.5 重复性
按“1.2.2”项下GM供试品溶液的配制方法,在相同条件下,平行制备6份样品,测定其峰面积,考察方法的重复性。
1.2.6 稳定性
取浓度为20μg/mL的GM对照品溶液和20μg/mL的供试品溶液,在室温条件下保存,分别于0、1、2、4和6h测定其峰面积,考察溶液的稳定性。
1.2.7 回收率
分别配制16、20和24μg/mL的GM对照品溶液,依次称取处方量的辅料至9个25mL的容量瓶中,分别加入高、中、低浓度对照品溶液,每种浓度分别制备3份,按供试品制备方法操作,取续滤液进样测量其峰面积,按对照品溶液峰面积计算GM的含量并求得其回收率。
1.3 处方制备
1.3.1 包合物的制备
参照文献[8-10]先称取原料药和包合材料用研钵进行研磨包合,再称取相关辅料,用等量递加的方式进行研磨混匀,以保证本品含量均匀度符合规定。
1.3.2 GM普通片及环糊精包合物片剂的制备
普通片制备通过过筛方式先将处方量的API与乳糖采用等量递加法过65目筛混合均匀,再依次加入其余辅料混合均匀,加入黏合剂制成软材,过24目筛制粒,湿颗粒在50℃干燥40min,过24目筛整粒,加入润滑剂混合均匀,调节装量在200mg左右,调节硬度60N左右,压片。GM包合物片剂制备采用研磨法先将处方量API与HP-β-CD加入研钵,充分研磨搅拌。然后根据等量递加法加入其他辅料研磨搅拌均匀,加入黏合剂制成软材,过24目筛制粒,湿颗粒在50℃干燥 40min,过24目筛整粒。取干颗粒加入润滑剂,混合均匀,调节装量在200mg左右,调节硬度60N左右,压片(原辅料均已提前过80目筛)。
1.4 处方优化
1.4.1 处方单因素实验
分别以不同的稀释剂比例、黏合剂种类、黏合剂浓度、崩解剂比例和润滑剂比例进行单因素实验,通过测定其累积溶出度使用f2因子比较不同处方间的差异(50≤f2≤100时,认为两条曲线相似或等同)。
1.4.2 响应面优化设计
通过单因素实验结果,以溶出度为响应值,稀释剂比例(A)、黏合剂浓度(B)和崩解剂比例(C)为因变量进行3因素3水平的BBD-RSM实验优化(表1)。通过Design Expert 8.0.6.1软件对数据进行结果分析,模型验证得出GM环糊精包合物的最优条件。
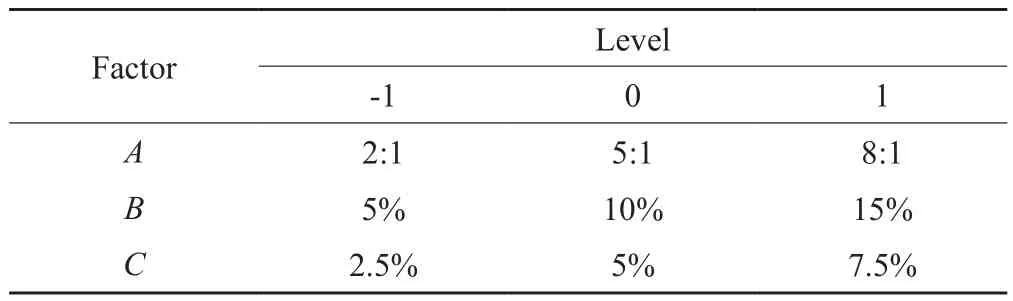
表1 BBD选择的变量编码及其水平Tab. 1 Level and code of variables chosen for box-behnken design
1.5 GM包合物验证
1.5.1 包合物的表征
用溴化钾压片法,对GM、HP-β-CD、GM/HP-β-CD物理混合物和GM/HP-β-CD包合物,分别在400~4000cm-1的波数范围进行红外吸收光谱测定。
1.5.2 溶解度的测定
称取过量的GM,GM/HP-β-CD物理混合物,GM/HP-β-CD包合物样品分别置于15mL试管中,加10mL水配成过饱和溶液,置于水浴恒温摇床振摇[(25±0.5)℃,120r/min] 48h,用0.45μm的微孔滤膜迅速过滤,将滤液经流动相稀释适当倍数后于228nm处由HPLC测定,代入标准曲线方程,经计算得到GM,GM/HP-β-CD物理混合物,GM/HP-β-CD包合物样品在水中的溶解度。
1.6 片剂的质量检查
按照中国药典四部[11]的要求依次对片剂的片重差异、脆碎度、溶出度及含量均匀度进行检查。
1.7 片剂的体外释放
采用重蒸水、pH7.8的磷酸盐缓冲液和0.02%的三羟基氨基甲烷为溶出介质,量取溶出介质900mL置于溶出杯内,待溶出介质温度恒定在37.0℃后,取6片供试品分别投入6个溶出杯内,设置转速75r/min,分别于5、15、30、60和90min时间点取样适量并补充相同体积的同温释放介质,用0.45μm滤膜进行过滤,取续滤液进高效液相色谱仪进行含量分析,计算累积溶出度。
1.8 统计学方法
响应曲面模型的回归方程式和显著性统计用Design Expert 8.0.6.1 软件进行计算和分析。单因素实验结果采用直观分析和方差分析,考察稀释剂比例、黏合剂种类、黏合剂浓度、崩解剂比例和润滑剂比例对其的影响。
2 结果与分析
2.1 HPLC法测定GC含量的方法学验证
标准曲线的回归方程为y=65248x+589.7(R2=0.9999,n=8),线性范围0.1~80μg/mL;精密度实验RSD为0.86%(n=6),重复性实验RSD为1.02%(n=6);稳定性实验对照品与供试品RSD分别为0.96%和1.21%;低、中和高3种剂量平均回收率分别为98.85%、100.63%和99.74%,RSD分别为1.19%、0.48%和0.85%(n=3)。结果表明,建立的HPLC含量测定方法符合试验测定要求。
2.2 单因素实验结果与分析
2.2.1 稀释剂比例的影响
使用初选处方辅料的稀释剂-乳糖和MCC,控制装量为200mg,按照2:1、5:1和8:1的比例配制处方,根据片剂的累积溶出度为考察指标进行筛选,结果见图1a。
结果表明,乳糖和MCC的比例对片剂的体外释放有显著影响,f2(2:1、5:1)和f2(2:1、8:1)分别为35.44和43.57。乳糖为水溶性辅料,MCC为水不溶性辅料,乳糖比例越大理论上溶出越快,但由于亲水性乳糖比例过大会导致片剂结合过于致密,反而会影响药物前期和中期的释放。
2.2.2 黏合剂种类的影响
使用初选处方辅料的黏合剂-淀粉浆、HPMC、PVP,考察其种类对片剂累积溶出度的影响,结果见图1b。
结果表明,黏合剂种类对片剂的体外释放有显著影响,f2(淀粉浆,PVP)和f2(PVP、HPMC)分别为25.15和32.11。淀粉浆作为一种传统使用的黏合剂,由于其糊化会导致药物包裹在其中,造成后期药物不易完全释放,因此在目前药物开发中使用的越来越少;HPMC由于在冷水中不易分散,而在热水中溶解性又较差,因此配制过程先要在热水中分散,随后冷却后再溶解,过程较为繁琐且在此处方中效果不及PVP。综合考虑,固化黏合剂为PVP。
2.2.3 黏合剂浓度的影响
通过筛选黏合剂种类,确定使用PVP为黏合剂,现进一步对其浓度进行筛选,现拟定5%、10%、15% 共3种浓度为比较对象,根据片剂的累积溶出度为考察指标进行筛选,结果见图1c。
结果表明,黏合剂浓度对片剂的体外释放有显著影响,f2(5%、15%)和f2(10%f15%)分别为34.94和34.42。使用15%PVP其溶出度较其他两种都低,且采用PVP 15%黏合剂制软材,黏合剂溶解较慢,后期释放较慢。而5%及10%的PVP总体差别不大,因此需进一步优化得出最佳处方。
2.2.4 崩解剂比例的影响
使用初选处方辅料的崩解剂-CMS-Na,考察其占处方总比例大小对片剂质量的影响。现拟定3种比例2.5%、5%和7.5%为比较对象,将片剂的累积溶出度作为考察指标进行筛选,结果见图1d。
结果表明,在拟定3种比例7.5%、5%、2.5%中,使用2.5%的崩解剂其溶出速率较其他两种都慢一点,f2(2.5%,5%)和f2(2.5%、7.5%)分别为39.60和37.50。随着崩解剂比例提高至5%,片剂的溶出体现出明显改善的趋势,但比例继续增大至7.5%,溶出的变化不是很明显,因此需进一步优化得出最佳处方。
2.2.5 润滑剂微粉硅胶的影响
使用初选处方辅料的润滑剂-微粉硅胶,考察其添加对片剂质量的影响。根据片剂的累积溶出度为考察指标进行筛选,结果见下图1e。
结果表明,微粉硅胶的比例对体外释放无显著影响,f2(0.5%、1%)和f2(1%、1.5%)分别为78.29和75.98。但考虑到添加微粉硅胶可以改善流动性且溶出度高,所压片剂硬度适宜,崩解时限也相对较快等特点;综合考虑固化微粉硅胶比例为1%。
2.3 响应面优化实验
2.3.1 BBD-RSM 实验设计结果
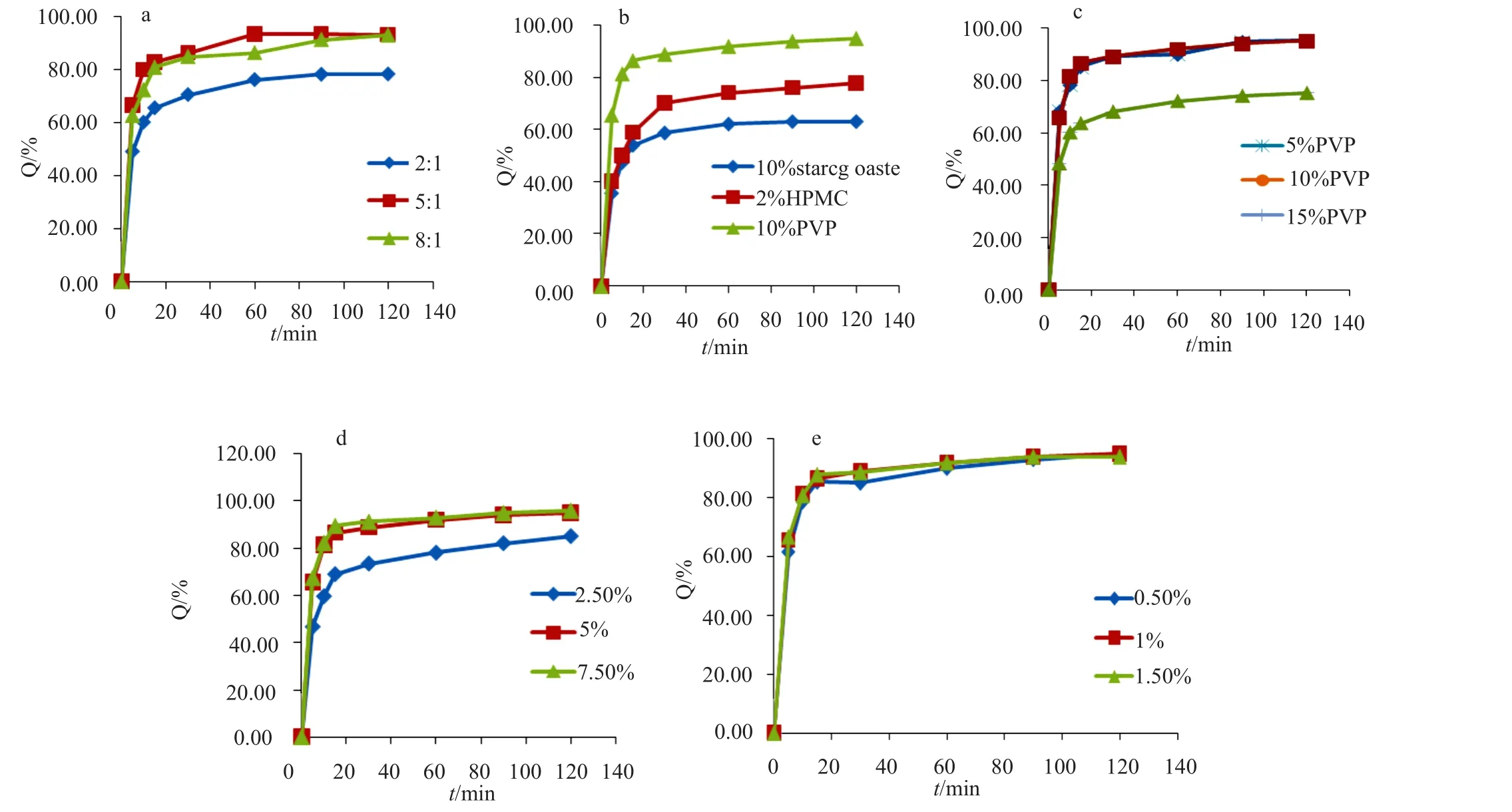
图1 不同影响因素的释放曲线Fig.1 Dissolution curves of different in fluencing factors
根据单因素实验结果表明:稀释剂比例(A)、黏合剂浓度(B)和崩解剂比例(C)是影响药物释放的主要因素,以其作为自变量,溶出度为响应值进行3因素3水平BBD-RSM实验优化(表1)。保持各处方API 2mg,黏合剂采用PVP,润滑剂占处方1%的量,用 Design-Expert 8.0.6进行实验设计,共17组处方组成,制备包合物片剂并测定各处方在15min的溶出度,结果见表2。
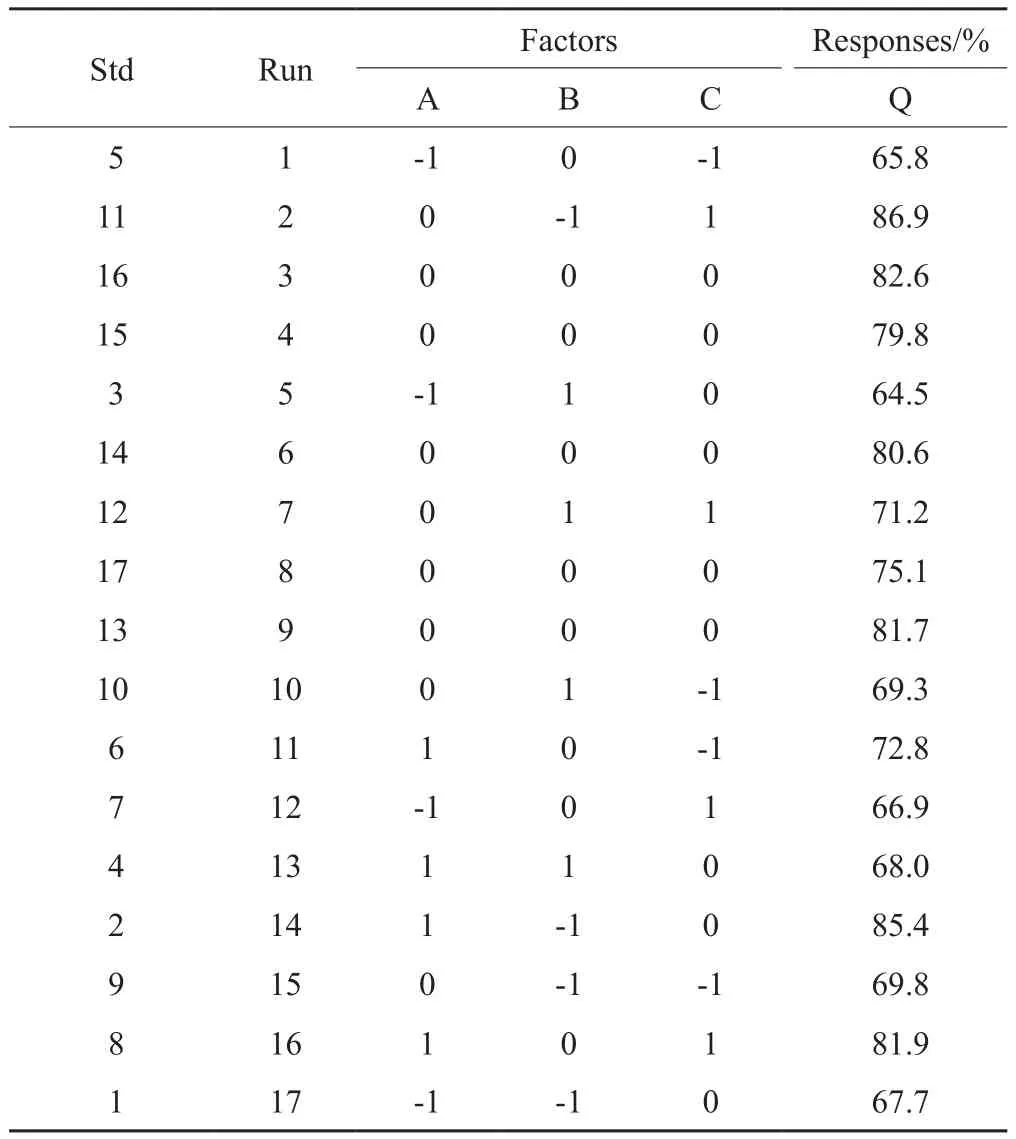
表2 BBD试验结果的实测值Tab. 2 Box-Behnken design matrix along with test values
2.3.2 响应面结果分析
经过模型拟合确定最优模型为二次多项式,模型具有显著性,失拟项无显著性,具有统计学意义。通过实验得到的模型回归方程为:Y=79.96+5.40A-4.60B+3.65C-3.55AB+2.00AC-3.80BC-5.51A2-3.06B2-2.60C2(R2=0.9483;模型:F=14.27,P=0.0010,有意义;失拟项:P=0.1636,无意义)。影响药物溶出的主次因素是:稀释剂比例(A)>黏合剂浓度(B)>崩解剂比例(C),除了因素A、B和C外,A2、B2以及交互项AB和BC都是影响溶出度的显著因素。包合物片剂溶出度的等高图及响应面3D图谱如图2所示。从图中可以看到,稀释剂乳糖和微晶纤维素的比例对片剂溶出度的影响最大,随着乳糖比例的提高,溶出度先急速增大,后又慢慢减小。其次,黏合剂浓度和崩解剂比例对溶出度也有较为显著的影响,随着黏合剂浓度提高,溶出度先缓慢增大然后显著减小;随着崩解剂比例增大,溶出度先升高后趋于平缓。通过直观图得出的结果与模型方程得到的结果一致,这也反映了模型方程的可靠。
2.3.3 优化条件的验证试验
采用 Design-Expert V8.0.6.1软件对方程中Y求解,得到HP-β-CD格列美脲片的理论最佳条件为:稀释剂-乳糖和MCC的比例为7.37:1,黏合剂-PVP的浓度为6%,崩解剂-CMS-NA的比例为6.45%。为了简化实际操作的难度,HP-β-CD格列美脲片最佳条件修正为稀释剂-乳糖和MCC 的比例为7.5:1,黏合剂-PVP的浓度为6%,崩解剂-CMS-NA的比例为6.5%。此条件下进行3组平行实验,所得溶出度的平均值为88.13%(n=3),而回归方程所得的溶出度理论预测值为88.69%,两者相对误差为0.64%,说明运用响应面法优化得到的模型参数准确可靠。
2.4 包合物的验证结果
2.4.1 红外吸收光谱
由图3所示,GM(a)在3369~3288cm-1为N-H拉伸振动的吸收峰(如3366和3291cm-1处的峰),在1708~1673cm-1处为格列美脲分子内的羰基伸缩振动吸收峰(如1706和1673cm-1处的峰),在1345~1153cm-1处为分子内磺酰胺基团的不对称伸缩振动吸收峰(如1340和1153cm-1处的峰);HP-β-CD(b)在3500~3300cm-1处为O-H拉伸振动峰(如3429cm-1处的峰),在3000~2800cm-1处为CH-CH2的伸缩振动峰(如2934cm-1处的峰);而HP-β-CD/GM物理混合物(c)的峰为a和b峰的简单叠加,GM和HP-β-CD的特征吸收峰均没有变化;HP-β-CD/GM包合物(d)的峰同a、b和c相比没有太大变化,说明骨架结果并没有发生变化,但在3369~3288cm-1处的峰明显加宽,且HP-β-CD在3429cm-1处的峰在(d)蓝移到了3348cm-1处,这是由于包合作用使GM的特征峰被HP-β-CD的峰减弱,同时HP-β-CD峰的环境改变导致振动频率降低。在a上1708~1673cm-1和1345~1153cm-1处的峰在d上只可观察到1345~1153cm-1处的峰,1708~1673cm-1处的峰在d上消失,这是因为形成包合物后,由于GM分子较大使得HP-β-CD只能部分包合还有一部分基团未被包合物包合进去,进一步说明确实形成了稳定的包合物。
2.4.2 水溶性测定
按溶解度的测定项下方法操作,经带入标准曲线后得出实验结果如表3所示。由表3可知,GM原料药的溶解度仅为1.36μg/mL,GM/HP-β-CD物理混合物的溶解度提高了20多倍,而GM/HP-β-CD包合物在水中的溶解度提高了50多倍。事实证明,GM被水溶性极好的HP-β-CD包合后,水溶性得到了极大的改善。
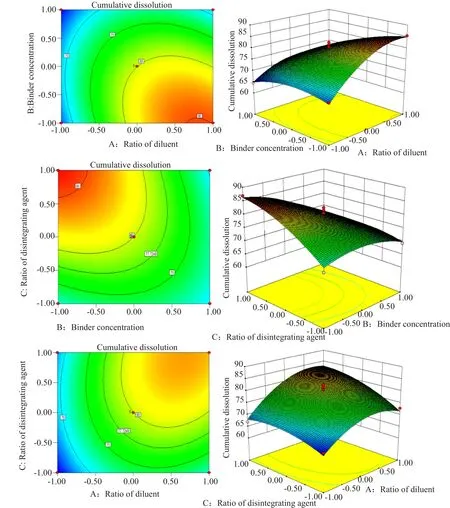
图2 影响因素对药物溶出度影响的等高线图与3D图谱Fig.2 contour map and 3D map of in fluencing factors on dissolution of drugs
2.5 片剂的质量检查结果
2.5.1 片重差异的检查
取供试品20片,精密称定总重量,求得平均片重后,再分别精密称定每片的重量,每片重量与平均片重比较,计算重量差异,结果见表4。
2.5.2 脆碎度的检查
取供试品32片洗耳球吹去粉末后称得总重,100转后洗耳球吹去粉末后再称得总重,计算减失重量,结果见表4。
2.5.3 含量均匀度的检查
取供试品1片置于50mL的容量瓶中,加1~2mL重蒸水,超声使药片崩解,加80%乙腈适量,使溶解后,定容摇匀,即得。用0.45μm的滤膜过滤后,取续滤液作为供试品溶液,进高效液相色谱仪分析其含量,结果见表4所示。
2.5.4 溶出度的检查
取本品依照中国药典(2015版)采用浆法测定GM片剂溶出,转速为75r/min,900mL pH7.8磷酸盐缓冲液(phosphate buffer, PBS)为溶出介质,15min取溶液过0.45μm的滤膜后取续滤液用HPLC分析,考察溶出度,结果见表4。
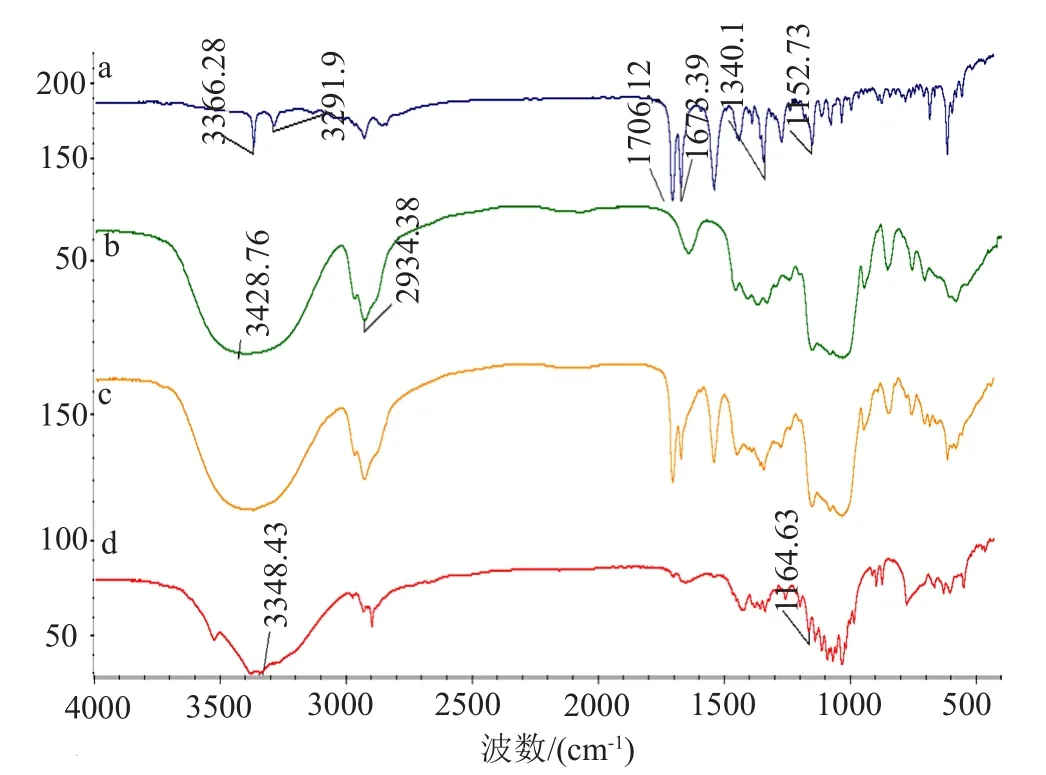
图3 GM(a)、HP-β-CD(b)、HP-β-CD/GM物理混合物(c)和HP-β-CD/GM包合物(d)红外光谱图Fig.3 Infrared spectra of glimepiride (a), HP-β-CD (b), HP-β-CD/glimepiride physical mixture (c), HP-β-CD/glimepiride inclusion complex (d)
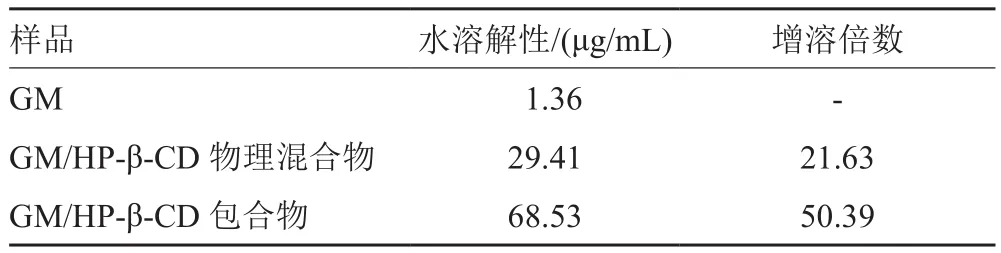
表3 样品的水溶性测定结果(n=3)Tab. 3 Determination of water solubility of samples (n=3)
由表4可知,制备的GM普通片及环糊精包合物片剂在含量均匀度、片重差异、脆碎度和溶出度方面均满足2015版药典要求。
2.6 体外释放曲线的绘制
采用重蒸水、pH7.8的磷酸盐缓冲液和0.02%的三羟基氨基甲烷为溶出介质,分别取格列美脲片和优化后的HP-β-CD格列美脲片,按片剂的体外释放项下方法进行含量测定,计算累积释放度,结果见图4。
由图4分析可知,在PBS和三羟基氨基甲烷的释放介质中,两种格列美脲片释放结果差异较小,90min时两者均已达到85%以上,相对较为稳定;而在水中两者的释放结果相差较大,90min时格列美脲片才达到60%,而HP-β-CD格列美脲片的释放已达93.28%,明显优于格列美脲片。作为难溶性药物,体外与体内释放具有一定的相关性,提升其体外释放意义重大;在0.02%的三羟基氨基甲烷溶液中,两者区分力最小,而在水中区分力最大,因此可以选择重蒸水作为筛选最优处方的释放介质。
3 讨论
HP-β-CD为水溶性材料,过大的溶解度导致使用饱和溶解度法不能很好地进行制备,而本文通过研磨法制备包合物具有操作简便、成本低廉和易于工业化生产等特点。通过响应面法对HP-β-CD格列美脲片处方进行优化,得到的优化模型预测性较好,其图形直观,且相比正交设计在实验条件寻优过程中,可以连续地对实验的各个水平进行分析。基于这些优势,响应面法将在今后的处方设计与优化过程中发挥越来越重要的作用。

表4 格列美脲片的质量检查结果Tab. 4 Quality controlled results of glimepiride table(n=3)

图4 处方在3种介质中的溶出曲线Fig.4 Dissolution curves of the prescription in three media
测定结果表明,HP-β-CD与GM形成的包合物成功地改善了溶解性,从而有望改善其生物利用度。在体外释放试验中,普通片和包合片剂在水溶液中的区分力较大而在其余两种介质中区分力较小,这是因为GM在0.02%的三羟基氨基甲烷溶液中的溶解度大,其溶解速率较快;而在pH7.8的磷酸盐缓冲液中,GM的溶解度仍是在水中的近30倍,加上两种片剂均提高了其溶解度,因此普通片与包合片剂并无大的区别;同理,在中性及酸性环境中,由于溶解度小,GM很依赖于制剂技术的改善。此前,由于检测条件的限制,规格较小的药物都采用小杯法进行溶出度的检测,但难溶性药物采用常规溶出介质很难达到漏槽条件,因此格列美脲片不得不采用三羟基氨基甲烷溶液这种专有介质,但这是以牺牲区分力为代价的。随着制剂技术的进步与HPLC的普及,微量的准确检测变得可能,除中国药典外,各国药典标准均无小杯法,是否停止采用小杯法以便同国际接轨,值得商榷。本文的研究结果将为GM制剂的开发奠定良好的理论基础。
猜你喜欢
中成药(2018年8期)2018-08-29 01:28:08
中成药(2018年6期)2018-07-11 03:01:40
中成药(2018年5期)2018-06-06 03:11:49
中成药(2018年1期)2018-02-02 07:19:55
中成药(2017年12期)2018-01-19 02:06:56
中成药(2017年10期)2017-11-16 00:50:08
中成药(2017年10期)2017-11-16 00:50:03
中成药(2017年5期)2017-06-13 13:01:12
特产研究(2016年3期)2016-04-12 07:16:16
中国卫生标准管理(2015年24期)2016-01-14 09:29:01
