
天然产物绵马酚B的合成研究
2019-01-30 06:49滕星星古金华王媛媛石培琪叶连宝
中国抗生素杂志 2019年1期
滕星星 古金华 王媛媛 石培琪 叶连宝
(广东药科大学药物化学教研室,广州 510006)
绵马酚B(aspidinol B)又名三叉蕨酚,是一种间苯三酚类化合物,主要分布于鳞毛蕨科(Dryopteridaceae)、鳞毛蕨属(Dryopteris Adanson)的粗茎鱗毛蕨(Dryopteris crassirhizomaNakai)和绵马贯众(Dryoperidis crassirhizomatisRhizoma)的根茎中,是这两种植物的特征化学成分[1],结构如图1所示。现代药理学研究表明,绵马酚B对白念珠菌,金黄色葡萄球菌,枯草芽孢杆菌,大肠埃希菌等都有着显著抗菌活性[2-3],对人类口腔表皮样癌细胞有选择性活性[4],有抗氧化作用[5-6],甚至可能成为治疗癌症的前沿药物。
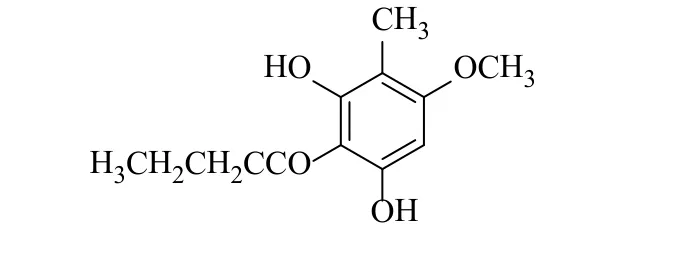
图1 绵马酚B的化学结构式Fig.1 Chemical structure of aspidinol B
目前报道中,有关于绵马酚提取方法很多,但仍存在缺陷,且目前尚未有行之有效的解决方法。例如,Toledano工作组[7]表明提取时会发生大分子的再聚合与自缩合现象。天然产物的合成早已成为如今合成化学家的重点趋势,但关于绵马酚B的合成文献却寥寥无几。
本实验室对早期Riedl的合成路线进行研究,对部分合成路线进行了改进[8]。为了避免酚羟基在后续反应过程中被氧化,原工艺采用苄基对酚羟基进行保护。考虑到苄基的毒性较大,本研究改用乙基碳化二亚胺/二甲基氨基吡啶(EDCI/DMAP)作为偶联剂保护酚羟基。首先采用了文献中的乙酸对羟基进行酯化(产率:43.71%),但后期实验发现异丁酸产率更高,故得到2-丁酰基-5-羟基-4-甲基间苯二甲酸二异丙酯(3)[9]。化合物(3)在碳酸钾的催化下与硫酸二甲酯反应约12h即可得到2-丁酰基-5-甲氧基-4-甲基间苯二甲酸二异丙酯(4),最后化合物(4)在碱性条件下水解成绵马酚B,总产率17.6%。与原路线中以2,4,6-三羟基甲苯为原料,经过傅克酰基化在2-甲基-间苯三酚(1)5位上加入丁酰基(产率44.64%),苄基氯保护酚羟基(产率:22.13%),硫酸二甲酯使羟基甲基化(产率:90.41%),最后用氢气脱保护(产率:51.62%)相比,改进后的路线操作简单,反应时间更短,总收率更高,更加环保,合成路线如图2所示,绵马酚B结构经鉴定与文献上描述一致[10-11]。
1 实验部分
1.1 主要仪器与试剂
RV8V-C旋转蒸发仪(德国IKA仪器公司);电热真空干燥箱(天津市华北实验仪器有限公司);XR6A显微熔点仪(上海精密仪器厂);Bruker Plus 400核磁共振波谱仪(日本日立公司);Quadrupoleu/MS型质谱仪(美国Perkin Elmer公司);Avance Ⅲ 400MHz核磁共振仪(CDCl3为溶剂,瑞士Bruker公司)。
实验所用试剂均为分析纯,购于广州化学试剂厂。
1.2 实验步骤
1.2.1 化合物2的合成
将2,4,6-三羟基甲苯3.50g(0.0250mol)置于250mL三口烧瓶中,加入二硫化碳16.0mL搅拌均匀后缓慢加入无水三氯化铝粉末10.0g(0.0750mol),随后在常温下沿瓶壁加入硝基苯9.40mL,加热回流0.5h后,将2.90g(0.0280mol)丁酰氯以及无水硝基苯1.60mL加入滴液漏斗中,于回流状态下缓慢滴入两者混合液,继续回流约1.5h后至反应完全冷却至室温,在搅拌下倾入盐酸冰水(浓盐酸15.0mL,冰95.0g)中水解20min,改为蒸馏装置加热用水蒸气蒸馏除去二硫化碳,趁热过滤,将滤液反复加热骤冷,析出浅黄色细针状结晶,合并所得结晶干燥后称重2.34g,产率44.64%。熔点162~165℃。1H NMR(400MHz,CDCl3) δ(ppm)6.11(s, 1H), 2.34(t,J=7.4Hz, 2H),2.16(s, 3H), 1.71(h,J=7.4Hz, 2H), 0.83(t,J=7.3Hz,3H)。13C NMR(100MHz, CDCl3) δ(ppm) 205.76(s),163.18(s), 159.95(s), 158.76(s), 110.43(s), 105.84(s),100.68(s), 46.30(s), 17.92(s), 13.73(s), 7.58(s)。EIMS: 212.56 [M+H]+。元素分析C11H14O4(210.23): 实测值(计算值),%:C 62.89(62.85); H 6.69(6.71); O 30.42(30.44)。
1.2.2 化合物3的合成
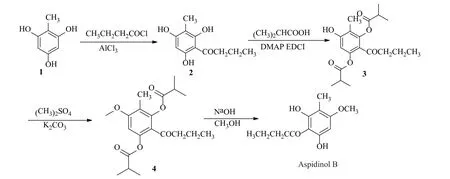
图2 绵马酚B的优化全合成方法Fig.2 The optimized synthetic method of aspidinol B
将6.00 g(0.0286 mol)化合物2,异丁酸5.33mL(0.0572mol),DMAP 0.342g(0.00280mol)溶于80.0mL THF,在-5℃下搅拌冷却;另取EDCI(16.4g,0.0860mol)溶于30.0mL的10%NaOH溶液中搅拌均匀,取80.0mL DCM进行萃取,然后将下层有机层滴加到上述溶液中去,保持零下温度持续反应3h,加水(1.00mL)淬灭反应,30℃减压蒸馏除去大部分THF,加水和DCM并用浓盐酸滴加到溶液中搅拌均匀将水层调至弱酸性,DCM萃取,无水硫酸钠干燥,将萃取液制砂,洗脱剂为乙酸乙酯/石油醚(体积比1:20) 进行柱层析分离,得黄色油状物6.36g,产率64.36%。1H NMR(400MHz, CDCl3) δ(ppm) 6.18(s,1H), 5.45(t,J=2.4Hz, 2H), 4.73(d,J=2.1Hz, 2H), 2.56(d,J=6.5, 2.7Hz, 2H), 2.32~2.28(m, 5H), 1.76~1.58(m, 2H),1.19(dd,J=6.8Hz, 1.3Hz, 12H), 0.92(t,J=7.5Hz, 3H)。13C NMR(100MHz, CDCl3) δ(ppm) 205.67(s), 158.18(s),155.93(d,J=14.6Hz), 153.67(s), 120.49(s), 105.20(s),97.48(s), 90.39(s), 55.78(s), 46.56(s), 30.06(s), 21.83(s),17.60(s), 13.92(s), 9.24(s). EI-MS: 351.32[M+H]+。元素分析C19H26O6(350.17):实测值(计算值),%:C,65.17(65.13); H, 7.53(7.48); O, 27.30(27.39)。
1.2.3 化合物4的合成
将5.00g(0.0144mol)化合物3溶于100mL丙酮,在常温条件下,加入硫酸二甲酯3.64g(0.0288mol),碳酸钾3.98g(0.0572mol),氮气保护,回流,反应12h后冷却,过滤除去白色钾盐,并用丙酮洗涤,减压条件下除去丙酮得黄色油状物5.13g,产率98.64%。1H NMR(400MHz,CDCl3) δ(ppm) 6.34(s, 1H), 5.67(t,J=2.1Hz, 2H),4.89(d,J=2.1Hz, 2H), 3.57(s, 3H), 2.78~2.46(m, 2H),2.25(t,J=7.4Hz, 2H), 2.21(s, 3H), 1.46(h,J=7.6Hz, 2H),1.45(dd,J=6.9Hz, 1.6Hz, 12H), 0.68(t,J=7.6Hz, 3H)。13C NMR(100MHz, CDCl3) δ(ppm) 205.36(s), 158.77(s),155.89(d,J=14.6Hz), 153.26(s), 112.46(s), 105.95(s),97.13(s), 90.47(s), 46.24(s), 30.87(s), 21.67(s), 17.24(s),13.69(s), 8.24(s). EI-MS: 364.82[M+H]+。元素分析C20H28O6(364.19): 实测值(计算值),%:C, 65.78(65.92);H, 7.33(7.74); O, 26.89(26.34)。
1.2.4 目标化合物伪绵马酚B的合成
将4.20g(0.0117mol)化合物4加入250mL圆底烧瓶中,加入甲醇60.0mL,2mol/L氢氧化钠水溶液60.0mL,氮气保护,40℃搅拌约2.5h,加入浓盐酸10.0mL中和,搅拌均匀后在减压条件下蒸馏除去甲醇,DCM萃取,萃取液干燥旋干,纯化采用洗脱剂为二氯甲烷/石油醚(体积比100:1)进行柱层析,得黄色固体2.97g,正己烷重结晶,得黄色针状固体1.62g,产率62.10%。1H NMR(400MHz, CDCl3)δ(ppm) 6.34(s, 1H), 3.79(s, 3H), 2.57(t,J=7.7Hz, 2H),2.48(s, 3H), 1.84(m, 2H), 0.26(t,J=7.3Hz, 3H)。13C NMR(100MHz, CDCl3) δ(ppm) 205.46(s), 168.59(s),158.75(s), 153.78(s), 112.93(s), 105.58(s), 100.33(s),61.78(s), 46.47(s), 17.58(s), 13.72(s), 8.66(s)。EI-MS:224.94[M+H]+。元素分析C12H16O4(224.10):实测值(计算值),%:C, 64.62(64.27); H,7.31(7.19); O,28.07(28.54)。
2 结果与讨论
通过对原合成路线的改进,本研究在原来14d左右的周期上缩短至3d,总产率从4.57%提升至17.6%,而化合物3和4未见文献报道。
在原路线中,化合物2使用毒性试剂卞基氯保护酚羟基保护苯环上2位以及4位酚羟基,经过6d反应在6位酚羟基甲基化,最后用10%钯碳脱去苄基保护基。改进后的路线,直接采用异丁酸保护化合物2-甲基-6-丁酰间苯三酚(2)的2位以及4位酚羟基,采用EDCI作为活化试剂,不仅可以吸收反应体系中少量的水,还可以防止反应过程中生成的活性中间体醚被破坏,DMAP的使用更是可以稳定活性中间体醚,延长其存在时间,阻止生成副产物,这样不仅减少了反应时间,更是提高了反应产率。在原路线中,反应时间最长的就是6位酚羟基甲基化,6d的反应时间导致整个反应路线周期的延长,而我们新合成路线中,由于苯环上2位和4位酚羟基成酯,增加了酚羟基的酸性,促进了硫酸二甲酯的甲基化,使化合物3经过12h反应完全,大大缩短了合成路线的周期。原路线采用了氢气还原去除苄氧基,此反应过程中由于氢气危险,需要使用高压釜进行反应。改进后的路线,由于前期使用了异丁酸进行酚羟基保护,在最后可以直接使用2mol/L氢氧化钠溶液对化合物4进行酸碱中和,即可在碱性条件下得到的目标产物绵马酚B。
总之,通过对原路线的改进,本研究得到了一条以2,4,6-三羟基甲苯为原料,经历4步反应即可得到绵马酚B的高效率优化合成路线,为之后aspidinol B的衍生物或药理学实验研究提供了更好的基础。
猜你喜欢
中国教育网络(2022年8期)2022-12-21
无机化学学报(2022年9期)2022-09-16
农业工程学报(2022年5期)2022-06-22
化工时刊(2022年1期)2022-05-25
城市道桥与防洪(2022年3期)2022-05-08
安全与环境工程(2021年2期)2021-04-02
煤炭加工与综合利用(2020年6期)2020-07-17
中国房地产业·下旬(2020年12期)2020-01-11
农产品加工(2019年19期)2019-10-23
电子制作(2018年23期)2018-12-26
