
大学生饮食习惯与唾液微生物多样性的关联
2019-01-28 06:09张国庆黄子琪王明月孔俊豪李余动陈建设
食品科学 2019年1期
张国庆,黄子琪,王明月,孔俊豪,李余动*,陈建设
(浙江工商大学食品与生物工程学院,浙江 杭州 310018)
人体微生物群是一个极其复杂的生态系统,以口腔微生物群为例,不同个体的口腔微生物群都会不同。这种微生物群组成和变化的多样性决定了人体口腔生态系统的平衡或失调,甚至决定着口腔疾病的发生和发展[1-2]。口腔微生物的种类保守估计有19 000 种[3],但目前口腔中能被检测分离出的微生物只有700 种,而且有近一半以上的细菌不可培养,如Anaerolineae、Bacteroidetes等在传统的微生物培养法中不能培养[4-5]。近年来,人们用分子生物学、分子生态学等手段对口腔微生物群落进行深入研究,发现很多因素,如饮食、人种及地域等均会引起口腔内微生物群落结构的差异[6]。Ren Wen等研究认为,口臭是由人体口腔微生物引起的,口臭患者的舌苔有更丰富的细菌[7]。口腔微生物既直接参与口腔疾病的发展,也通过与其他人体微生物的交互作用间接影响人体健康。Atarashi等的研究发现,炎症性肠病患者唾液中的肺炎克雷伯氏菌会迅速定植到小鼠肠道,导致强烈的炎症反应,直接证实了口腔细菌与肠道疾病之间的关系[8]。
Illumina MiSeq是新一代高通量测序平台之一,该测序平台克服了传统测序方法读长较短、误差较大等缺陷,因而可获得更准确的微生物群落信息[9-10]。16S rRNA基因全长约1 542 bp,在结构和功能上都具有高度的保守性,且存在于所有的细菌中,因此常常被用于微生物鉴定。16S rRNA基因的9 个高变区(V1~V9)中,V4区是最准确且能识别到属水平的可变区,而V3~V4区测序读长比V4区更长,相较单独V4区测序更加准确[11-12]。
据调查,大学生群体对口腔健康的知识了解较少,口腔的健康状况不容乐观[13],尤其是饮食习惯对大学生口腔健康的影响应该引起足够重视。本实验采用Illumina MiSeq测序平台,基于16S rDNA V3~V4高变区对不同饮食习惯的大学生群体的唾液微生物进行研究,以期揭示饮食习惯对唾液微生物群落结构及其多样性差异的影响,促进在校学生对合理膳食与口腔健康的重视。
1 材料与方法
1.1 实验对象与试剂
实验采样对象为在读大学生,通过问卷的形式对对荤素饮食、是否饮酒、是否喝茶和咖啡等含咖啡因饮料,以及是否运动等10余项差异选项进行调查。调查结果显示以荤素饮食差异为研究方向较好,群体区分较其他项明显,最终在尽可能平衡口腔卫生等影响因素的条件下,选取符合实验要求的大学生36 名,依据每周饮食摄入荤食时间分成2 组:每周摄入荤食时间不少于4 d为A组,少于4 d为B组[14]。选取标准:1)取样前3 个月无抗生素使用史,龋、失、补牙数小于2,无其他口腔疾病;2)无先天性疾病及系统性疾病(如艾滋病、肝炎、糖尿病等),6 个月内无重大疾病史;3)受试者取样前12 h内未实施口腔保健(如刷牙、嚼口香糖等),取样前6 h内无进食、饮酒、喝含咖啡因的饮料等。该研究提供唾液样品的大学生均悉知实验目的并签署知情同意书。
DNA Mini Kit 德国QIAGEN公司;Premix Ex TaqTMHot Start Version 日本Takara公司。
1.2 仪器与设备
聚合酶链式反应(polymerase chain reaction,PCR)仪美国Bio-Rad公司;Illumina MiSeq测序平台 美国Illumina公司。
1.3 方法
1.3.1 采样方法
唾液采集采用自然流出法,具体方法为,用一次性无菌取样杯收集志愿者自然流出、清澈无痰的唾液,采样量大于2 mL。采样过程注意避免污染,唾液样品直接用于后续实验,避免唾液样本低温保存对结果造成影响[15-16]。
1.3.2 DNA提取
使用DNA Mini Kit提取唾液样品中总DNA,提取方法严格按照试剂盒说明书进行,提取得到的DNA于-80 ℃冻存,备用。
1.3.3 16S rRNA基因扩增及高通量测序
提取得到的D N A样本1 0 0 μ L,对D N A的V3~V4区进行PCR扩增[17](使用引物序列为338F:5’-ACTCCTACGGGAGGCAGCAG-3’;806R:5’-GGA CTACHVGGGTWTCTAAT-3’[18-19])。使用的聚合酶为Premix Ex TaqTMHot Start Version。电泳检测合格的PCR扩增产物送至检测机构,用Illumina MiSeq测序平台双端测序(Paired-end)分析。
1.4 数据分析
1.4.1 质量控制
测序所得原始数据,采用QIIME软件(v1.9.1)的微生物16S rRNA分析流程,对原始数据进行质量控制,舍弃低质量序列(Q<20),通过barcode区分样品序列,去除primer序列,进行overlap连接,去除杂合体,最终获得用于分析的序列,每个样本的序列超过10 000 条认为数据符合要求。
1.4.2 群落结构分析
采用QIIME软件将序列聚类成可操作分类单元(operational taxonomic unit,OTU),并对所有样本的序列按0.97的相似度进行OTU聚类,选取每一类中最长的序列作为OTU的代表序列,使用Greengene数据库对OTU代表序列进行物种注释,得到每个OTU的分类学信息[20]。统计各个样品包含的OTU情况及每个OTU中含有的序列的数目。采用QIIME进行组间差异分析,将属水平上的分类信息分别按照样品和分组进行聚类,做出热图(heatmap图)。采用QIIME进行主成分分析(principal component analysis,PCA),其中一个点代表一个样品,颜色相同的点属于同一分组,2 个点之间的距离越近,表示2 个样品中的微生物群落差异越小。
1.4.3 统计学分析
使用统计软件R对各组样品进行单因素方差分析[21],P<0.05表示组间差异具有统计学意义。
2 结果与分析
2.1 唾液细菌基因组DNA提取

图1 V3~V4区PCR结果Fig.1 Agarose gel electrophoresis of PCR products of bacterial 16S rRNA V3-V4 region
通过对比试剂盒法(DNA Mini Kit)与化学法(吐温提取法)对部分唾液样品的细菌DNA提取的影响,发现采用化学法提取的DNA电泳拖尾现象严重(泳道9~12),提取浓度也不稳定,综合对比后认为DNA Mini Kit的提取效果较化学法好(图1),DNA纯度与浓度均达到构建测序文库的要求,最后采用此试剂盒提取细菌DNA用于测序。最新研究也表明,唾液样品的不同收集方法与DNA提取方法对唾液微生物组成的检测影响并不大[22-23]。
2.2 唾液细菌种群的丰度与多样性
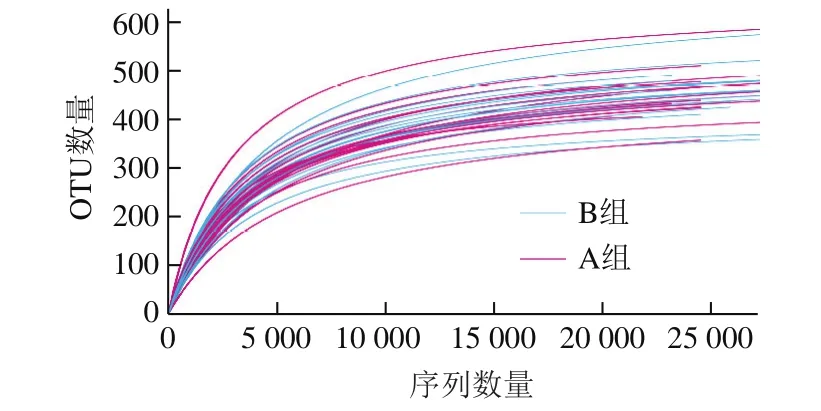
图2 样品稀释曲线Fig.2 Rarefaction curves
本实验共得到990 286 条优质序列(每个样品平均27 508 条),发现了212 个物种,在门和属2 个水平上对同类的OTU进行聚类,其归属于16 个门、143 个属。各样品的稀释曲线均趋于平缓(图2),表明测序已趋于饱和,测序深度已基本覆盖样品中所有物种。

表1 多样性指数Table1 Diversity indices
本实验中样本α多样性分析中的多样性指数如表1所示。其中A组和B组Simpson多样性指数分别为0.93±0.04与0.95±0.02,Shannon多样性指数分别为5.54±0.63与5.78±0.03,2 组样本有着相似的多样性指数,所有样本的覆盖度均大于95%。
2.3 唾液细菌种群结构
2.3.1 门水平分布
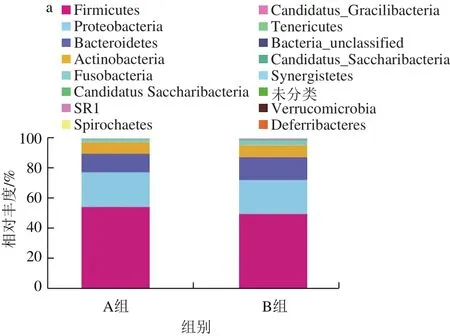

图3 样本的门水平菌群丰度(a)及优势菌(b)Fig.3 Abundance of salivary bacteria (a) and dominant bacteria (b) at the phylum level

表2 不同分类水平上样品的细菌群落结构及其相对丰度Table2 Bacterial community structure and their relative abundance at different taxonomic levels%
本实验检出的微生物共涉及16 个门,2 组样本共有的优势菌门(菌群丰度>1%)为Firmicutes、Proteobacteria、Bacteroidetes、Actinobacteria、Fusobacteria,其中Firmicutes、Proteobacteria、Bacteroidetes、Actinobacteria不存在显著性差异,而Fusobacteria存在极显著性差异(P<0.01)(图3)。在非优势菌门中,SR1、Saccharibacteria在A组的相对丰度显著低于B组(P<0.05)(表2)。
2.3.2 属水平分布

图1 样本的属水平菌群丰度(a)及优势菌(b)Fig.1 Abundance of salivary bacteria (a) and dominant bacteria (b) at the genus level
本实验共检出11 3 个属,在优势属(图4)中,A组中Streptococcus相对丰度比B组极显著增加(P<0.01),Fusobacterium相对丰度比B组极显著减少(P<0.01),而在非优势属中,A组中Peptostreptococcus、Parvimonas、Eubacterium、Saccharibacteria_genera_incertae_sedis、SR1_genera_incertae_sedis、Catonella、Mogibacterium、Solobacterium相对丰度均比B组显著减少(P<0.05),A组中Corynebacterium相对丰度比B组显著增加(P<0.05)(表2)。
2.4 样品的PCA与聚类分析
2.4.1 PCA分析
采用QIIME进行PCA分析,其中一个点代表一个样本,相同的颜色表示是同一组的样本,2 个点之间的距离越近,说明2 个样品的微生物群落差异越小。由PCA结果可知,2 组样本均比较分散,主成分区分度不是特别明显,但在各自象限均占主要优势(图5),说明2 组样本的主成分有一定差异,但没有显著差异。这可能与本研究的采样对象均为健康大学生群体有关,还需要扩大取样人群范围进一步研究。

图5 A组和B组样品的主成分分析Fig.5 Principal component analysis of salivary microbial communities in two groups
2.4.2 聚类分析
物种分类heatmap图用颜色变化来反映二维矩阵或表格中的数据信息,它可以直观地将数据值的大小以定义的颜色深浅表示出来。将属水平上的分类信息进行组间丰度相似性聚类,将聚类后数据表示在热图上,将高丰度和低丰度的物种分块聚集,通过颜色梯度及相似程度来反映样品在各分类水平上群落组成的相似性和差异性。

图6 A组和B组样品的菌群组成热图Fig.6 Heat map analysis of salivary microbial community structures in two groups
由菌群组成的热图(图6)可知,红色代表细菌丰度高,蓝色代表细菌丰度低,2 组样本的菌落组成存在差异,如Haemophilus、Gemella、Rothia等在A组中丰度较高,Peptostreptococcus、Neisseria、Saccharibacteria、Fusobacterium等在B组中丰度较高,而Streptococcus在2 组中没有明显变化。
3 讨 论
学者们普遍认为口腔疾病的发生和发展是由口腔中微生物群落结构所调控,而其中唾液微生物有重要的影响[24-25]。随着高通量测序技术的发展以及微生态的兴起,人们越来越关注口腔微生物群落的结构和多样性对健康的潜在影响[26]。从微生物群落结构差异的角度来探索个人饮食习惯与健康的关联是微生态研究的切入点。
本研究关注大学生这一群体,实验研究对象选择在校大学生,饮食与生活习惯相对一致,且学校生活较为规律,其他环境因素影响较小,所选用的个体均经过严格筛选,尽可能选择在其他方面统一的志愿者,保证样本的可靠性。通过对荤素饮食不同的两组大学生口腔内微生物群落结构的分析发现,两组样本有着相似的菌群多样性指数,但部分菌群的丰度,在门水平和属水平上存在显著组间差异;其中,还有值得关注的潜在致病菌或条件致病菌,如Corynebacterium、Streptococcus等菌属[27-28]。
口腔唾液有着复杂的微生物群落结构,本研究结果表明口腔唾液中Corynebacterium、Streptococcus等潜在致病或条件致病菌的属与常吃荤食呈正相关,而Peptostreptococcus、Eubacterium等无害菌属与常吃荤食呈负相关。这种微生物种群差异也说明,饮食习惯对口腔微生物存在一定影响,合理饮食结构,多吃蔬菜水果,控制肉类的摄入,将有助于人们的身体健康[29]。需要指出的是,本研究结果还需要后续加大样本数量,设置重复样品,并通过收取志愿者不同时间周期的样品[30],研究口腔的微生物菌群动态变化,进一步研究饮食习惯与菌群结构变化的关系,对于指导在校学生合理膳食具有现实意义。
猜你喜欢
世界科学技术-中医药现代化(2022年9期)2023-01-17
当代水产(2022年8期)2022-09-20
中老年保健(2022年2期)2022-08-24
中国饲料(2022年5期)2022-04-26
昆明医科大学学报(2022年3期)2022-04-19
昆明医科大学学报(2022年2期)2022-03-29
食品安全导刊(2021年20期)2021-08-30
中国生殖健康(2020年8期)2021-01-18
科学(2020年4期)2020-11-26
河南科学(2020年3期)2020-06-02
