
迷迭香黄酮的微波提取及其β-环糊精钾金属有机骨架材料包结物稳定性研究
2019-01-26 07:44聂吉语姜子涛常金涛李逸凡
食品工业科技 2019年1期
聂吉语,姜子涛,李 荣,王 颖,常金涛,李逸凡
(天津商业大学生物技术与食品科学学院,天津市食品生物技术重点实验室,天津300134)
迷迭香(Rosmarinus officinalis L.)属唇形科迷迭香属植物[1],别名海洋之露、艾菊。迷迭香原产于环地中海沿岸地区,近年在我国云南、贵州、海南等地已被大面积引种栽培[2]。迷迭香多年来一直被用作香料,用于佐餐及腌制、熏制食品的调味[3]。对于迷迭香化学成分的研究,国内外已有许多文献报道[4-6],其成分主要包括黄酮类、萜类和精油等[7-9]。其中,二萜类物质迷迭香酸具有抗辐射、抗诱变、保护脑组织和治疗过敏性疾病等药理学活性[10-11]。另外,陈宝等[12]和江涛等[13]分别报道了迷迭香黄酮的提取工艺,包括乙醇热浸提法及有机溶剂法等;Hyun等[14]测定了迷迭香甲醇提取物的乙酸乙酯提取部分的抗氧化能力,并从中鉴定出了咖啡酸、迷迭香酸、迷迭香酸甲酯、木樨草素、芹黄素和高车前素6种成分。类似地,陈文等[15]利用LC-MS/MS分离鉴定了迷迭香中的5种化学成分。已报道的迷迭香黄酮提取方法包括有机溶剂法、索氏提取法、回流提取以及超声辅助提取等[16-18],这些方法均存在提取时间长、提取效率低等缺陷。
本研究采用微波辅助快速提取的方式[19]提取迷迭香黄酮,通过单因素实验和响应面法确定迷迭香黄酮的最佳提取条件,并利用大孔吸附树脂对所获得的迷迭香黄酮进行提纯;采用β-环糊精钾金属有机骨架材料(K-CD-MOF)对迷迭香黄酮进行包结,优化其在加热、强酸或强碱等特殊加工环境下的稳定性,以期为迷迭香黄酮的加工利用提供参考。
1 材料与方法
1.1 材料与仪器
迷迭香 粉碎后过20目筛备用,购自云南;D101大孔树脂 天津南开大学化工厂;芦丁标准品 北京化学试剂公司;甲醇(色谱纯) 天津市科密欧化学试剂有限公司;无水乙醇、石油醚(30~60℃) 天津市南开区依文司乐玻璃器皿销售中心;亚硝酸钠、硝酸铝 天津金汇太亚化学试剂有限公司;氢氧化钾、氢氧化钠、冰乙酸 天津市凯通试剂有限公司;β-环状糊精(β-CD) 上海化学试剂采购供应站联营企业中心化工厂;所用试剂除特殊注明外,均为分析纯;超纯水 实验室自制。
1260 Series型高效液相色谱仪(色谱柱为Agilent Zorbax Eclipse Plus-C18,250 mm × 4.6 mm,5μm) 美国 Agilent公司;Multisynth型微波反应器 意大利Milestone公司;FW100型高速万能粉碎机 天津市泰斯特仪器有限公司;Lambda 25型紫外-可见分光光度计 珀金埃尔默仪器有限公司;Alpha-1500型紫外可见分光光度计 上海谱元仪器有限公司;RE52-86A型旋转蒸发仪 上海亚荣生化仪器厂;FD5-3型冷冻干燥机 美国GOLD SIM公司;B203型生物光学显微镜 重庆市奥特仪器有限公司;NICOLET iS10型傅里叶变换红外光谱仪 美国Thermo Nicolet公司;AUW120D型十万分之一天平 日本岛津公司;SMART-N型纯水机 上海Heal Force公司。
1.2 实验方法
1.2.1 标准曲线的制作及黄酮含量的测定 参考文献[20]的方法绘制黄酮的标准工作曲线,精确称量20.0 mg的芦丁标准品,用体积分数为30%的乙醇溶液溶解后,转移到100 mL容量瓶中并定容,即得质量浓度为0.2 mg/mL的芦丁标准溶液。取上述芦丁标准溶液0.0、0.5、1.0、1.5、2.0、2.5 mL 分别置于10 mL 比色管中,依次加入0.4 mL 5%NaNO2溶液,摇匀,静置6 min后加入0.4 mL 10%Al(NO3)3溶液,摇匀,静置6 min后加入4.0 mL 4%NaOH溶液,摇匀,最后用体积分数为30%的乙醇溶液定容至刻度。以试剂空白为对照,15 min后于508 nm波长处测其吸光度。得到标准曲线回归方程为:A=10.27143C+0.01783(R2=0.9995),式中,C为芦丁的质量浓度(mg/mL),A为吸光度值。
按公式(1)计算黄酮得率:
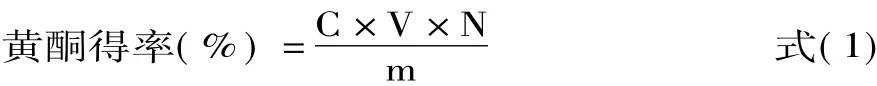
式中:C为提取液由芦丁标准曲线计算出的黄酮质量浓度(mg/mL),V为提取液体积(mL),N为稀释倍数,m为称取的迷迭香粉末的质量(mg)。
1.2.2 迷迭香黄酮微波提取工艺 称取1.0 g脱脂后迷迭香粉末,放入微波反应器的平底烧瓶中,以一定体积分数的乙醇溶液为溶剂,设置一定的微波功率、微波温度以及提取时间,于微波反应器中提取。冷却后减压过滤,所得滤液用石油醚萃取3次,弃除石油醚层,得迷迭香黄酮提取液。经减压旋蒸至无乙醇后冷冻干燥得迷迭香黄酮粉末。
1.2.3 迷迭香黄酮微波提取单因素实验 选择液料比40∶1(mL∶g)、微波温度70℃、微波功率500 W以及提取时间7 min,分别考察乙醇体积分数在20%、30%、40%、50%、60%、70%、80%时的黄酮得率;在乙醇的体积分数为50%的溶液为提取溶剂,微波温度70℃、微波功率500 W以及提取时间7 min,分别考察液料比在20∶1、30∶1、40∶1、50∶1、60∶1(mL∶g)时的黄酮得率;选择体积分数为50%的乙醇溶液为提取溶剂,液料比40∶1(mL∶g),微波功率500 W和提取时间7 min,分别考察微波温度在40、50、60、70、80℃时的黄酮得率;选择体积分数为50%的乙醇溶液为提取溶剂,液料比 40∶1(mL∶g),微波温度70℃,分别考察微波功率在400、500、600 W下,提取时间在5、6、7、8、9 min时的黄酮得率。所获得的提取液在最大吸收波长273 nm下测定其吸光度A,以吸光度值的大小表征提取液中黄酮的得率的高低。试验重复3次,取平均值。
1.2.4 迷迭香黄酮提取工艺的响应面优化设计 根据单因素实验结果及SPSS软件分析,选取乙醇体积分数(A)、液料比(B)、提取温度(C)作为响应面设计的考察因素并选取较优水平范围,以迷迭香黄酮在273 nm处的吸光度值为响应值,按照 Box-Behnken原理,用Design Expert 8.0设计优化试验。其中,每个因素3个水平,中心试验重复3次,因素水平设计如表1。
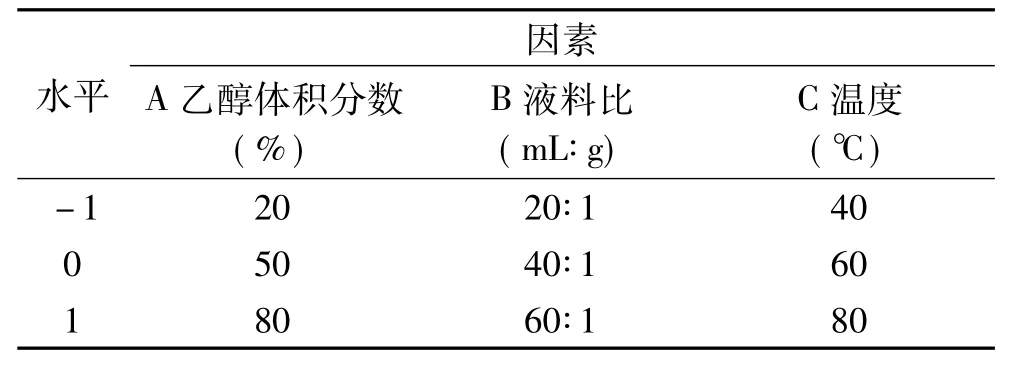
表1 响应面试验因素水平设计Table 1 Factors and levels design of response surface methodology
1.2.5 迷迭香黄酮样品液的制备 称取迷迭香黄酮粉末0.5 g,加入少量无水乙醇溶解,转移至500 mL容量瓶中,蒸馏水定容,即得1.0 mg/mL的迷迭香粗黄酮储备液。
1.2.6 D101大孔树脂纯化迷迭香黄酮
1.2.6.1 大孔树脂的预处理 参考文献[21]中的方法并稍作修改。将D101大孔树脂足量浸泡于体积分数为95%的乙醇溶液中24 h,装柱,用蒸馏水洗至流出液无乙醇味,然后用5%的HCl溶液浸泡3 h,装柱,用蒸馏水洗至流出液pH呈中性,再用5%的NaOH溶液浸泡3 h,装柱,用蒸馏水洗至流出液pH呈中性。将处理好的大孔树脂浸泡在蒸馏水中,备用。
1.2.6.2 D101大孔树脂纯化迷迭香黄酮 称取适量经预处理的D101大孔树脂,采用湿法装柱。参考文献[22],调节1.0 mg/mL迷迭香粗黄酮储备液p H为4,流出液速度控制在2 BV/h,每隔8 min测定一次在273 nm波长下流出液的吸光度值,当流出液的吸光度值为上样液吸光度值的1/10(泄漏点)时,停止上样,此时达到大孔树脂的最大吸附量。用2 BV蒸馏水洗净柱内残留样品液以及水溶性杂质,然后用50%乙醇溶液解吸,解吸液体积为3 BV、流速为2 BV/h,收集迷迭香黄酮解析液。用旋转蒸发仪减压浓缩后冷冻干燥,得纯化后迷迭香黄酮粉末,按公式(2)计算纯化后黄酮纯度。
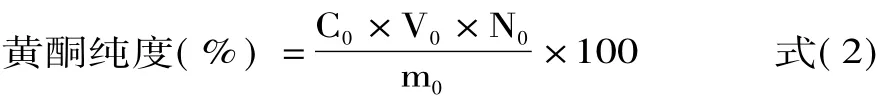
式中:C0为纯化后迷迭香黄酮的质量浓度(mg/mL);V0为迷迭香黄酮溶液的体积(mL);N为稀释倍数;m0为迷迭香黄酮粉末的质量(mg)。
1.2.6.3 HPLC法检测D101大孔树脂对迷迭香黄酮纯化效果 将纯化前后迷迭香黄酮粉末分别用色谱级甲醇溶解,配制成质量浓度为1.0 mg/mL的溶液,进行HPLC分析。色谱条件:流动相为甲醇(A)、超纯水(B)、1%乙酸水溶液(C)三者的混合溶液,采用梯度洗脱的方式:在0~45 min之间,A的浓度由30%匀速增加到80%,B由60%匀速降低到10%,期间C的浓度始终保持10%;流动相流速为0.8 mL/min;进样量10μL;检测波长273 nm;柱温30℃。
1.2.7 K-CD-MOF的合成及对迷迭香黄酮的包合作用
1.2.7.1 K-CD-MOF的合成 参照文献[23-24]的方法并稍作改进,准确称取1.3000 gβ-CD和0.4500 g氢氧化钾于烧杯中,加入20 mL水磁力搅拌溶解,全部溶解后将烧杯放入装有100 mL无水甲醇的层析缸中,密封。在室温条件下静置一周,期间甲醇蒸汽缓慢扩散进入溶液,在烧杯中生长出无色长立方块晶体K-CD-MOF。用玻璃棒取少量合成得到的K-CD-MOF材料,平铺在显微镜的载玻片上,观察其形貌。
1.2.7.2 迷迭香黄酮K-CD-MOF包结物(RF-K-CD-MOF)制备 参考文献[25]的方法,按 1∶3、1∶5、1∶8、1∶10 和1∶12 的芯壁比(以质量比计)进行试验。即称取一定量的K-CD-MOF,用蒸馏水将其溶解,在48℃下向K-CD-MOF溶液中逐滴滴加纯化后迷迭香黄酮溶液,边滴加边搅拌1 h,待包结物冷却后,冷冻干燥即得不同芯壁比下的包结物干燥粉末。
1.2.7.3 包结率测定 参考文献[26]的方法测定包结率。包结物表面黄酮的测定:称取各芯壁比的RF-K-CD-MOF粉末0.0500 g于100 mL容量瓶中,加入无水乙醇,充分振荡、洗涤后,无水乙醇定容至刻线,摇匀后静置,移取1.0 mL上清液,按照亚硝酸法显色[20],测定 RF-K-CD-MOF表面黄酮的吸光度,并按方法1.2.1的芦丁标准曲线计算黄酮含量,实验平行做3次。
RF-K-CD-MOF中总黄酮的测定:称取各芯壁比的RF-K-CD-MOF粉末0.0500 g于100 mL容量瓶中,加水定容至刻线,60 W超声溶解包结物,静置,移取1.0 mL上清液,按照亚硝酸法显色,测定RF-K-CD-MOF中总黄酮的吸光度,并按方法1.2.1的芦丁标准曲线计算黄酮含量,实验平行做3次。
则RF-K-CD-MOF中黄酮包结率按公式(3)计算:

1.2.7.4 包结物的红外光谱表征 分别将纯化后迷迭香黄酮(RF)、K-CD-MOF、RF-K-CD-MOF 及纯化后迷迭香黄酮与K-CD-MOF混合物和干燥后的KBr混合研磨均匀,在波数4000~500 cm-1进行红外光谱扫描。
1.2.8 RF-K-CD-MOF稳定性的研究
1.2.8.1 RF-K-CD-MOF的热稳定性 分别取RFK-CD-MOF及适量迷迭香黄酮粉末,均置于70℃恒温箱中保存,每隔24 h取样并配制成溶液,测定黄酮含量变化,并按公式(4)计算保存率。

1.2.8.2 p H对RF-K-CD-MOF稳定性的影响 分别配制p H为3.0、7.0、10.0的磷酸缓冲液,备用。称取RF-K-CD-MOF及适量迷迭香黄酮粉末,用50%乙醇配成溶液,分别加入等体积pH=3、p H=7和p H=10的缓冲溶液,于室温下保存,每隔24 h取样测定黄酮含量变化并按1.2.8.1的方法计算保存率。
1.3 数据处理
用SPSS对单因素实验数据进行分析,用Design-Expert对响应面实验数据进行分析。
2 结果与分析
2.1 单因素对黄酮提取的影响
根据1.2.3实验方法,固定乙醇体积分数、液料比、微波温度、微波功率与提取时间等五个单因素中的四个因素,分别考察了乙醇体积分数、液料比、微波温度、微波功率与提取时间各单一因素对总黄酮得率的影响,结果见图1。通过SPSS软件对单因素实验数据分析处理,结果见表2。

表2 单因素实验方差分析Table 2 Variance analysis of single-factor experiment
由图1a的结果可知:随着乙醇体积分数的增加,吸光度也不断增加,表明总黄酮得率不断增加。当乙醇体积分数增大到50%时,吸光度达到最大,此时再增大乙醇体积分数,吸光度反而逐渐减小,说明提取溶剂的极性既不能太低也不能太高。当乙醇体积分数过小时,提取溶剂极性较大,会溶出许多非目标物质,如水溶性的糖类等,并且为后续分离纯化黄酮带来不利影响[27]。当乙醇体积分数过大时,脂溶性的物质如叶绿素被溶出[28],且高体积分数的乙醇溶液会使细胞内的蛋白质凝固[29],多方面的因素都会影响黄酮的溶出,故提取迷迭香黄酮的最佳乙醇体积分数在40%~60%之间。
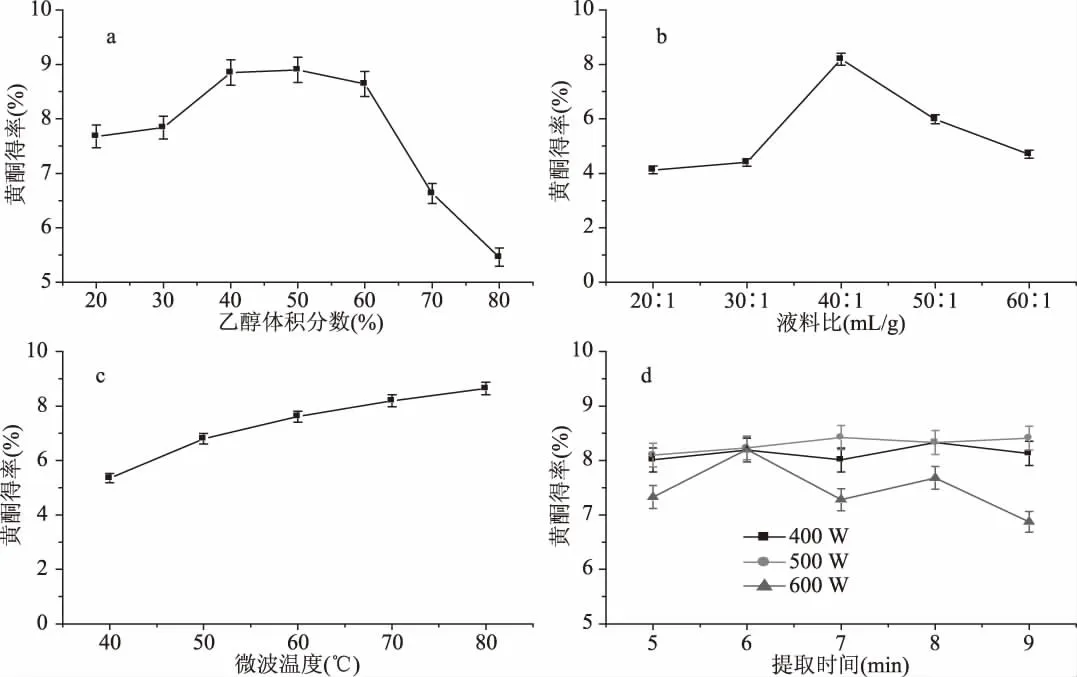
图1 各因素对迷迭香黄酮得率的影响Fig.1 Effects of various factors on the extraction rates of total flavonoids of rosemary
由图1b的结果可知:随着液料比的增加,吸光度也不断增加,表明总黄酮得率不断增加,液料比的增大会在很大程度上提高传质推动力[30]。当液料比增大到40∶1(mL∶g)时,吸光度达到最大,说明此时提取溶剂的量已经能充分溶解迷迭香中的黄酮。再增大液料比,吸光度反而逐渐减小,这是由于液料比过大会使混合溶液浓度变小,造成溶剂的浪费,故提取迷迭香黄酮的最佳液料比为40∶1(mL∶g)。
由图1c的结果可知:在一定微波功率下,随着微波温度的升高,吸光度也不断增加,表明总黄酮得率不断增加。当微波温度在40~80℃之间时,吸光度值有较明显的变化,表明总黄酮得率逐步增加。温度的升高会使提取溶剂的表面张力和黏性降低,使得分子震动更剧烈,从而使溶剂的渗透力以及对黄酮的溶解能力增加,有利于提高黄酮的提取效率;温度越高,达到最大得率的时间就越短。然而,温度持续升高,也会在不同程度上破坏黄酮类化合物的结构。综合考虑,提取迷迭香黄酮的最佳温度确定为60℃。
由图1d的结果可知:在一定温度下,同一时间微波功率为500 W的吸光度大于400 W和600 W的吸光度。由微波反应器的原理[31]可知,微波功率主要影响升温速率,当设定的微波温度一定时,微波功率越大,达到设定温度所需的时间就越短,达到设定温度后,微波反应器会根据所设定的温度自动调整微波功率,即在剩下的提取时间中用较低的微波功率维持所需的温度。因此不同微波功率会对应不同的最佳提取时间,并在此时间下达到总黄酮的最大提取得率。实验表明,当微波功率在500 W,提取时间在7 min时吸光度达到最大,即总黄酮提取得率达到最大,故提取迷迭香黄酮的最佳微波功率在500 W左右,最佳提取时间在7 min左右。
由表2可知:各因素对总黄酮得率的影响依次为:乙醇体积分数>微波温度>液料比>微波功率>提取时间。因此选取对总黄酮得率影响更显著的乙醇体积分数、微波温度和液料比作为响应面优化试验的因素。
2.2 响应面试验设计及结果
根据2.1的实验结果,在下一步的响应面试验中,考察乙醇体积分数(A)、液料比(B)、微波温度(C)这3个主要影响因素,将微波功率设定为500 W,提取时间设定为7 min。实验方案及结果见表3。
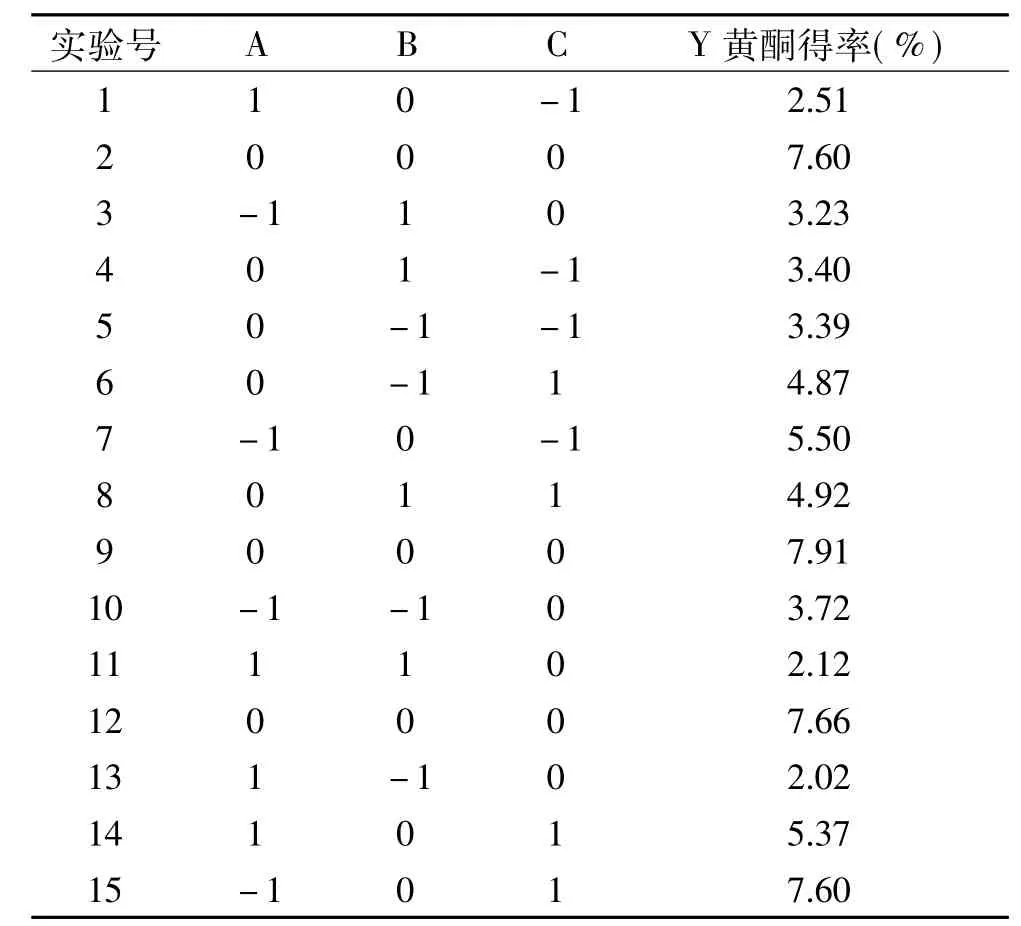
表3 响应面试验设计及结果Table 3 Response surface design and results
2.3 回归方程的建立及方差分析
用Design Expert对表3中的数据进行多元回归拟合,得到以吸光度Y为响应值,以乙醇体积分数A、液料比B、微波温度C为自变量的回归方程为:Y=0.81-0.078A-3.125×10-3B+0.077C+0.012AB+0.015AC+7.500×10-4BC-0.15A2-0.23B2-0.043C2,回归模型方差分析结果以及回归方程系数显著性检验结果如表4所示。
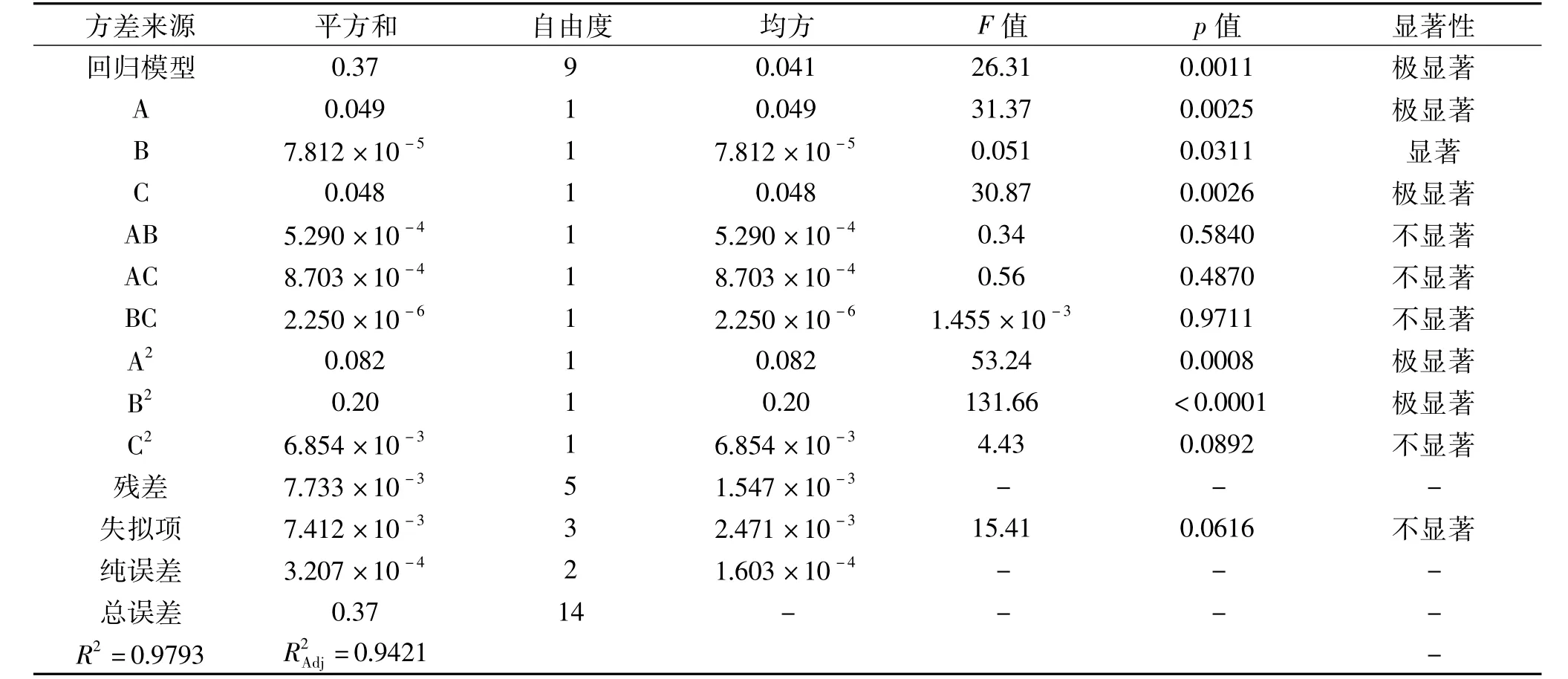
表4 回归模型的方差分析Table 4 ANOVA for the regression model
对表4中数据进行分析可以得到,整个回归模型是极显著的(p<0.01),失拟性检验不显著(p=0.0616>0.05),表明该模型拟合性较好,模型的调整确定系数R2=0.9783,说明该模型能解释97.83%响应值的变化,因此可用该模型对迷迭香总黄酮的提取进行分析和预测。影响因素中A、C、A2、B2极显著(p<0.01),B 显著(p<0.05),C2及交互项 AB、AC、BC对响应值吸光度Y的影响不显著,各因素对吸光度值影响的顺序为:乙醇体积分数>微波温度>液料比。
2.4 响应面分析
响应曲面图是响应值(吸光度值Y)对各个试验因素A、B、C所构成的三维空间曲面图形,从响应面图中可以直观地看出各个试验因素及两两因素之间的交互作用对响应值的影响。根据所得回归方程,以273 nm处的吸光度值Y作为响应值,分别将乙醇体积分数、液料比、提取温度定为0水平,绘制出三维响应曲面图及相应等高线,如图2。
由图2a可见,在乙醇体积分数和液料比的协同作用下,当乙醇体积分数在43%左右,液料比在40∶1(mL∶g)左右时吸光度达到最大,与2.1节单因素实验结果相符。
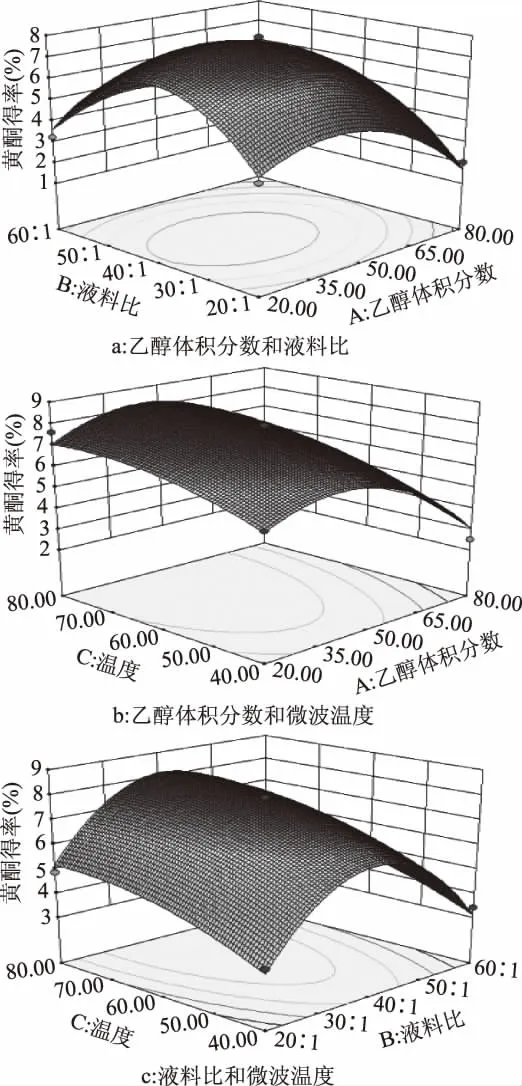
图2 各因素交互作用对吸光度的影响Fig.2 Effects of interactions of various factors on absorbance
由图2b可见,在乙醇体积分数和微波温度的协同作用下,当乙醇体积分数在43%左右,微波温度在77℃左右时吸光度达到最大。二者协同作用时,乙醇体积分数越大,沸点越接近乙醇的沸点(78.4℃),因此当温度达到80℃附近时,能观察到回流管中有大量的液体,即溶剂已达到沸点开始剧烈的沸腾,影响黄酮的溶出,使吸光度减小。
由图2c可见,在微波温度和液料比的协同作用下,当微波温度在77℃左右,液料比在40∶1(mL∶g)左右时吸光度达到最大。吸光度随着微波温度的升高以及液料比的增大而增大,二者的协同作用体现在微波温度较高时,会使溶剂的黏性降低,并使溶剂分子的振动频率增大,更多的溶剂能渗透进植物,此时较大的液料比就能更充分的溶解黄酮,使吸光度增大。
2.5 最佳提取条件的确定
通过响应面试验优化得到迷迭香总黄酮提取的最佳工艺条件为:乙醇体积分数为43.45%,液料比为39.78∶1(mL∶g),温度为77.18 ℃,微波功率500 W,提取时间7 min,该模型在此条件下,吸光度的预测值为0.851,总黄酮得率为8.26%。但考虑到试验实际可操作性,将迷迭香总黄酮提取的最佳工艺条件修正为:乙醇体积分数45%、液料比40∶1(mL∶g)、温度77℃。对修正的最佳工艺条件做验证试验,经过3次平行实验,吸光度平均值为0.821,总黄酮得率为7.88%,与预测值较接近,相对误差为4.6%。由此可见,该优化条件提取迷迭香总黄酮是可靠的。超声辅助提取迷迭香总黄酮可大大缩短提取时间,由文献[16]中报道的回流提取迷迭香总黄酮所需的4 h缩短至7 min,很大程度的提高了提取效率。
2.6 D101大孔树脂对迷迭香纯化效果分析
参照文献[21]的方法对迷迭香总黄酮进行纯化,由公式(2)得其纯化后纯度由30%提高到70%。并利用HPLC法对其纯化效果进行检测,图3为经D101大孔树脂纯化前后迷迭香总黄酮样品液在273 nm下的高效液相色谱图。从图3中可以看出,迷迭香总黄酮经D101大孔树脂纯化后的响应值比同浓度下纯化前的有明显增大,纯化后样品的响应值均明显增大2.3倍左右,这将为后续的成分分析、抗氧化等试验奠定一定基础。由此可知,D101大孔树脂对迷迭香黄酮类化合物能起到很好的纯化作用。
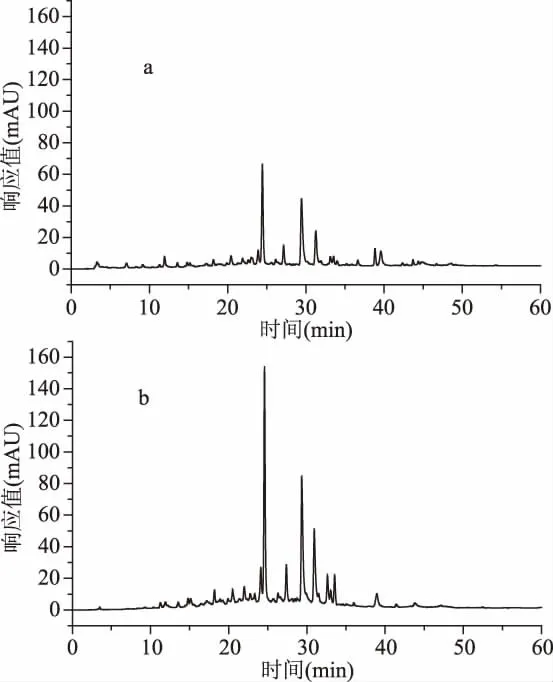
图3 D101大孔树脂纯化前后迷迭香总黄酮在273 nm下的HPLC图Fig.3 HPLC profile of the total flavonoids of the rosemary monitored at 273 nm by D101 resin
2.7 K-CD-MOF晶体形态的显微镜观察
图4显示了按照方法1.2.7.1合成得到的K-CD-MOF材料的显微镜照片,可见制备出的K-CDMOF材料属于立方晶体,晶型完整且表面光滑。该络合物的傅里叶变换红外光谱(FTIR)见图6。

图4 K-CD-MOF材料的显微镜照片(40×)Fig.4 Microscope image of K-CD-MOF(40×)
2.8 迷迭香黄酮K-CD-MOF包结物的包结率
按照方法1.2.7.2和1.2.7.3进行实验,计算得到不同芯壁比包结物的包结率,结果如图5所示。由图5可知,随着壁材(K-CD-MOF)的增加,迷迭香黄酮包结物的包结率增大;当芯壁比达到1∶10时,包结率达到87.9%,而当芯壁比达到1∶12时,包结率并无明显增大。因此,采用迷迭香黄酮与K-CD-MOF材料制备RF-K-CD-MOF的最佳配比确定为1∶10。
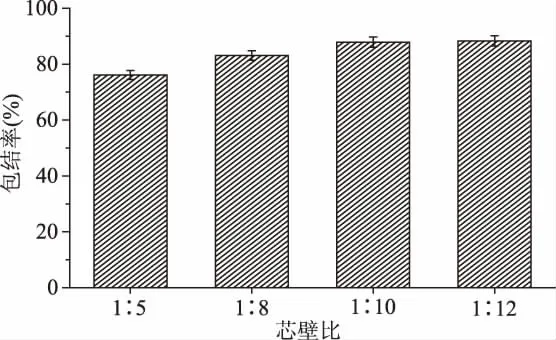
图5 不同芯壁比时RF-K-CD-MOF中黄酮的包结率Fig.5 Flavonoids inclusion rate of RF-K-CD-MOF in different ratio of core to wall
2.9 RF-K-CD-MOF的红外光谱测定
RF、K-CD-MOF、RF-K-CD-MOF 及其混合物的红外光谱图见图6。从图6中可以看出,RF与KCD-MOF形成包合物后其红外光图谱类似于两者的混合物图谱。根据文献[32-35]可知,3200~3500 cm-1为-OH的特征吸收峰,2930 cm-1处为-CH的特征吸收峰,1690 cm-1处为-C=O 的特征吸收峰,1450~1620 cm-1为芳环的特征吸收峰,1020~1190 cm-1为C-O及 C-O-C 伸缩振动吸收峰,900 cm-1处为 β-糖苷键的特征吸收峰。在3280 cm-1及1020 cm-1处,包合物的峰强度明显大于RF和K-CD-MOF,然而二者的混合物在此处的峰强度并未发生明显变化;包合物保留了RF中-C=O和芳环的特征吸收峰以及K-CD-MOF的空腔结合,但由于包合作用的发生,在1370和1020 cm-1等处峰有明显偏移,且RF中部分峰已被钝化,由此可推断出RF-K-CD-MOF包合物形成。从K-CD-MOF的FTIR图中可以看到,在3280、2922、1690、1370、1190、1020 和 885 cm-1等处出现了特征吸收峰,由此可以证明K-CD-MOF络合物的形成,并且保留了CD的空腔结构。
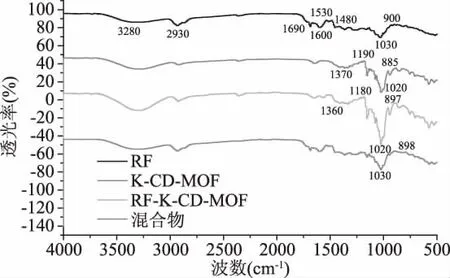
图6 迷迭香黄酮、K-CD-MOF、RF-K-CD-MOF及其混合物的红外光谱图Fig.6 IR spectra of RF,K-CD-MOF,RF-K-CD-MOF and the mixture
2.1 0 RF-K-CD-MOF的热稳定性及酸碱稳定性评价
2.1 0.1 加热对迷迭香黄酮及其包结物稳定性的影响 根据方法1.2.8.1进行实验,测得迷迭香黄酮及其包结物在70℃恒温条件下贮存一段时间后有效成分含量变化结果,如图7所示。从图7可以看出,相同环境下,经过1 d加热,迷迭香黄酮提取物的总黄酮保存率降至49.5%,而其包结物中总黄酮的保存率仅降至89.99%;维持70℃贮存9 d后,RF-KCD-MOF中总黄酮的保存率仍有60%以上,而提取物中总黄酮的保存率仅有20%,即总黄酮保存率从20.9%提高到60.6%。因此,采用K-CD-MOF材料对迷迭香黄酮进行包埋,可以有效地提高迷迭香黄酮提取物粉末的热稳定性。
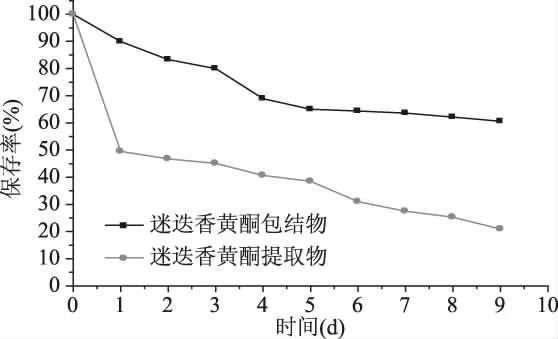
图7 加热对迷迭香黄酮及其包结物稳定性的影响Fig.7 Effects of heat on stability of rosemary flavonoids and its inclusion complex
2.1 0.2 pH对迷迭香黄酮及其包结物稳定性的影响 根据方法1.2.8.2进行实验,得到pH对迷迭香黄酮及其包结物稳定性的影响,结果如图8所示。结果表明,酸性条件下,迷迭香黄酮及其包结物中总黄酮成分的稳定性较好,而强碱性环境下极其不稳定,据文献报道[8],迷迭香中含有迷迭香酸,侯建春等[36]研究表明迷迭香酸的稳定性在pH3~6条件下要强于在pH7~9条件下。在中性条件下迷迭香总黄酮稳定性不好,而RF-K-CD-MOF表现出很好的稳定性,总黄酮保存率从41.69%提高到82.58%。这是因为迷迭香黄酮含有许多的多羟基酚类化合物,它们在弱酸性环境(pH4~6)中可以较稳定的以分子形式存在,这种状态有利于其在环糊精空腔中稳定存在[37-38]。因此,总体上看,RF-K-CD-MOF 具有较好的p H稳定性,且较适宜在酸性至中性条件下应用。
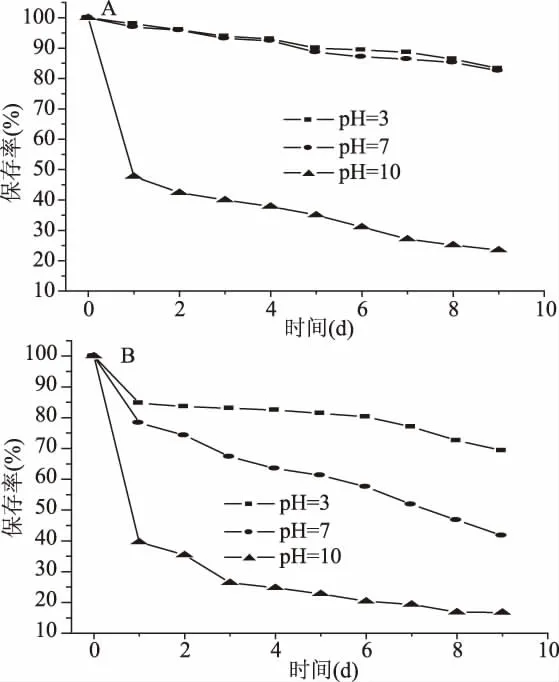
图8 pH对RF-K-CD-MOF稳定性的影响Fig.8 Effects of pH on stability of rosemary flavonoids and its inclusion complex注:图8(A)为RF-K-CD-MOF中黄酮的保存率变化;图8(B)为迷迭香黄酮提取物中黄酮的保存率变化。
3 结论
通过单因素实验确定迷迭香总黄酮微波提取各条件的范围,并通过响应面法优化得到提取的最佳工艺条件:乙醇体积分数45%,液料比40∶1(mL∶g),温度77℃,微波功率500 W,提取时间7 min,在此条件下的迷迭香总黄酮得率为7.88%;采用D101大孔树脂纯化迷迭香黄酮,确定上样液p H=4、流速2 BV/h、解析液为50%乙醇溶液,以及解析流速为2 BV/h,纯化后总黄酮纯度高达70%,为纯化前2.3倍。
制备了以一种新型的材料K-CD-MOF为壁材的迷迭香黄酮微胶囊,最大包结率达到87%以上。迷迭香黄酮提取物及其包结物的热及酸碱稳定性实验表明:采用K-CD-MOF材料对迷迭香黄酮进行包埋,可以有效地提高其热稳定性及酸碱稳定性。这种由可食用的β-CD和对人体无害的钾金属合成的新型K-CD-MOF材料,有望成为一种被广泛应用于食品工业中的绿色材料。这一研究为迷迭香黄酮在食品工业中的应用提供理论依据。
猜你喜欢
电子测试(2022年16期)2022-10-17
陶瓷学报(2020年6期)2021-01-26
汽车电器(2019年1期)2019-03-21
中成药(2018年10期)2018-10-26
制造技术与机床(2018年10期)2018-10-13
科技视界(2018年22期)2018-10-08
幼儿教育·教育教学版(2017年10期)2017-12-13
中国光学(2015年5期)2015-12-09
中国铸造装备与技术(2012年5期)2012-11-04
中国铸造装备与技术(2012年4期)2012-09-01
