
X连锁中性粒细胞减少症1例报告并文献复习
2019-01-25 10:23:44刘俐嫔刘璐瑶刘丹如孙金峤王晓川
中国循证儿科杂志 2018年6期
孟 新 侯 佳 王 莹 刘俐嫔 刘璐瑶 刘丹如 孙金峤 王晓川
1 病例报告
男,6岁5月,发现粒细胞减少6年余,为求进一步诊治,于2018年4月就诊于复旦大学附属儿科医院(我院)临床免疫科。
既往史:患儿3月龄时因发热查血常规,首次发现WBC及中性粒细胞绝对计数(ANC)减少,其后6年间断随访血常规,WBC及ANC始终低于正常水平。患儿平素易出现无明显诱因发热,频率约每月1次,热峰39℃左右,热型不规则,对症处理后发热可缓解;反复发生上呼吸道感染、化脓性扁桃体炎、口腔溃疡,感染病原包括白色念珠菌、EB病毒、肺炎支原体。先后使用多种抗感染药物,包括阿奇霉素、美罗培南、哌拉西林他唑巴坦、头孢菌素类(包括头孢哌酮他唑巴坦、头孢唑肟、头孢他啶、头孢唑林、头孢尼西),抗感染治疗后病情好转。3岁时于外院行骨髓细胞形态学检测,提示骨髓增生活跃低水平,粒系成熟障碍。曾注射重组人集落刺激因子(G-CSF),效果不详。无乙肝、结核、伤寒等传染病史及其接触史,有芒果过敏史,无药物过敏史,无手术外伤史,无异物吸入史。
个人史:G1P1,足月顺产,出生体重3.2 kg,Apgar评分1、5 min均为10分。生后母乳喂养,4月龄起添加鸡蛋黄、粥等辅食,现普食,无挑食、偏食。2个月会抬头,6个月会坐,13个月会走,智力、体格发育同正常同龄儿。
预防接种史:未按时接种,就诊前已全部补种,未见不良反应。
家族史:父母体健,非近亲婚配,否认家族性遗传性疾病史。
体格检查:T 36.5℃,P 100·min-1,R 22·min-1,体重21 kg。神志清楚,精神可,发育正常,营养中等。全身皮肤未见皮疹、出血点及黄染。浅表淋巴结未及肿大。外耳道无分泌液,乳突区无压痛。唇红,口腔黏膜光滑,咽部充血,双侧扁桃体I°肿大,充血,无脓苔。呼吸平稳,双肺呼吸音粗,未闻及啰音。心率100·min-1,律齐,心音有力。腹平软,未及包块,肝脾肋下未触及,肠鸣音2~3·min-1。四肢末端温,脊柱、四肢无畸形。克氏征、布氏征、巴氏征阴性。
实验室检查:2017年8月17日至2018年5月30日共行21次血常规检查,其中WBC(2.15±0.5)×109·L-1,ANC(0.261±0.086)×109·L-1,患儿于2018年4月29日至5月30日每2周随访血常规,ANC未见周期性波动(图1)。淋巴细胞亚群分类计数示NK细胞减少(23.42×109·L-1,1.63%)。骨髓细胞学染色(图2):骨髓有核细胞增生活跃低水平,粒红比为1.4∶1;粒系增生伴成熟障碍,部分细胞内可见颗粒增粗或可见空泡;红系及淋巴系细胞增生活跃,形态无明显异常;巨核细胞易见,血小板不少,簇状易见。
根据患儿临床表现,ANC减少、骨髓细胞形态学特点,临床怀疑先天性中性粒细胞减少症。外院血液遗传疾病基因筛查,提示WAS基因突变,致病性不确定。为进一步明确诊断,经患儿家长同意,对患儿及其父母进行WAS基因检测。
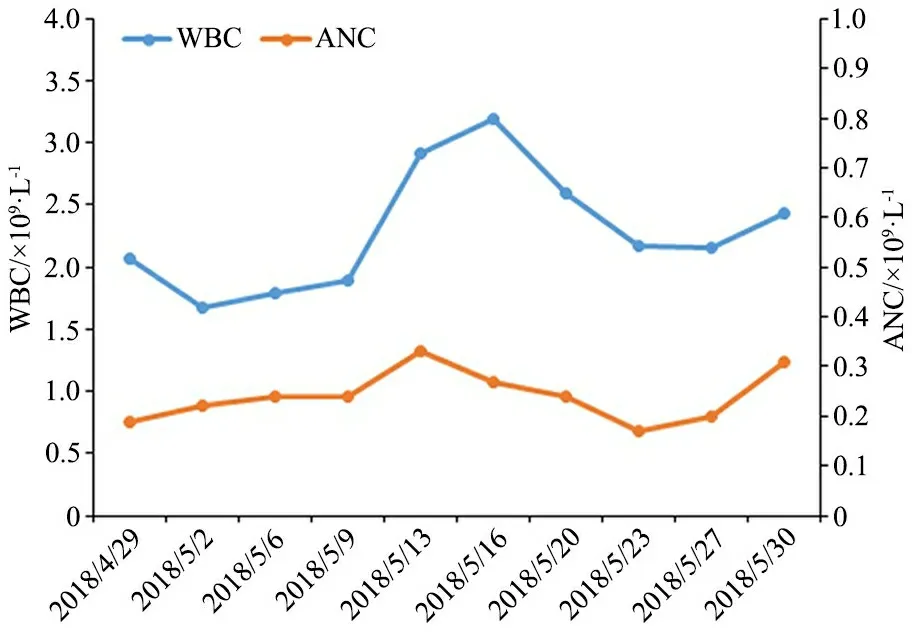
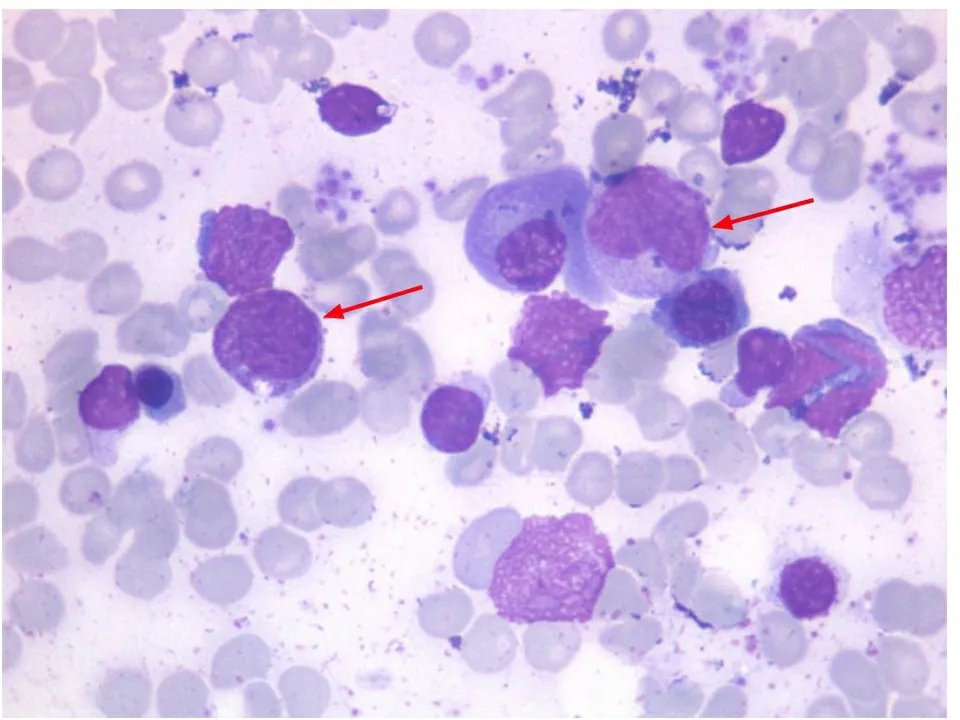
图1 WBC和ANC波动图
图2骨髓细胞形态学染色(瑞氏染色,×1000)示粒系成熟障碍
注 红色箭头示早幼粒细胞和中幼粒细胞
WAS基因Sanger测序验证:取患儿及父母新鲜肝素抗凝血各2 mL,用FlexiGene DNA Kit抽提全血基因组DNA,测DNA浓度约100 ng·μL-1,设计WASexon8-exon9上下游PCR扩增引物,PCR扩增WAS基因,HiSeq2000进行Sanger测序,FinchTV软件对结果进行分析(图3)。患儿X染色体9号外显子c.T881C(p.I294T)半合子变异,其母为携带者,其父该位点未发现变异。
WASmRNA检测:取患儿及父母新鲜肝素抗凝血各2 mL,分离PBMC(人外周血淋巴细胞分离液,购自天津灏洋生物),Trizol法提取总RNA并检测RNA浓度后,逆转录成cDNA(PrimeScriptTMRT Master Mix,Takara),设计qPCR引物(Forward Primer 5’-AGGTCGGAGTCAACGGATTT-3’; Reverse primer 5’-TTCCCGTTCTCAGCCTTGAC-3’),qPCR采用Takara TerraTMqPCR Direct SYBR®Premix推荐方案,Roche Light Cycler实时荧光定量PCR仪检测患儿及其父母WAS基因mRNA表达水平。以患儿父亲为正常对照,将其qPCR结果2-△△Ct设为1,母亲(携带者)为正常对照的0.60倍,患儿仅为正常对照的0.29倍,表明WAS基因p.I294T变异导致患儿该基因转录相对减少,携带者母亲的mRNA表达水平亦下降。
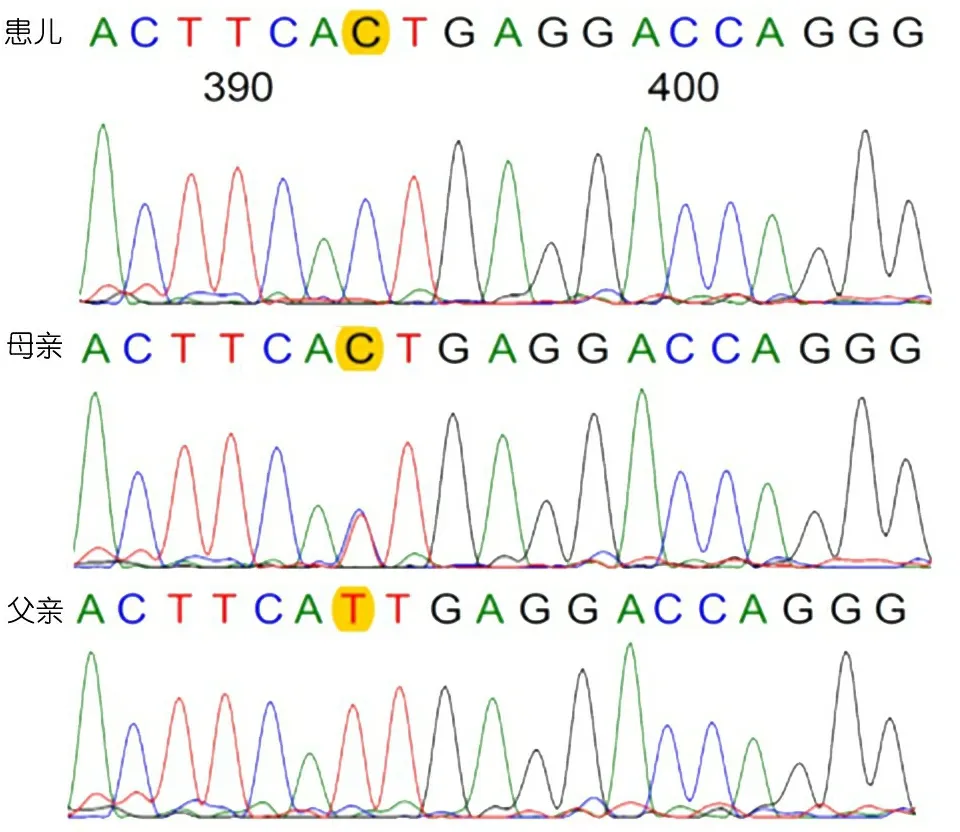
图3家系Sanger测序结果
注 黄色标记显示患儿c.T881C:p.I294T半合子变异,其母为携带者,其父该位点未发现变异
流式细胞术检测WASP表达:取患儿、患儿母亲及健康人新鲜血各1管,肝素抗凝分离PBMC,FACS buffer洗涤后与CD3抗体4℃孵育20 min,FACS buffer洗涤,加入250 μL FACS Staining Buffer重悬混匀,加入500 μL Cytofix/Cytoperm置于4℃反应30 min,Perm/Wash液洗涤,50 μL Perm/Wash液重悬后加入WASP抗体,4℃反应20 min,Perm/Wash液终止染色并洗涤,200 μL FACS buffer重悬后流式细胞仪检测WASP表达,FlowJo 软件进行数据分析。图4显示,患儿、母亲及正常对照细胞内WASP表达水平分别为74.5%、84.3%和93.7%。
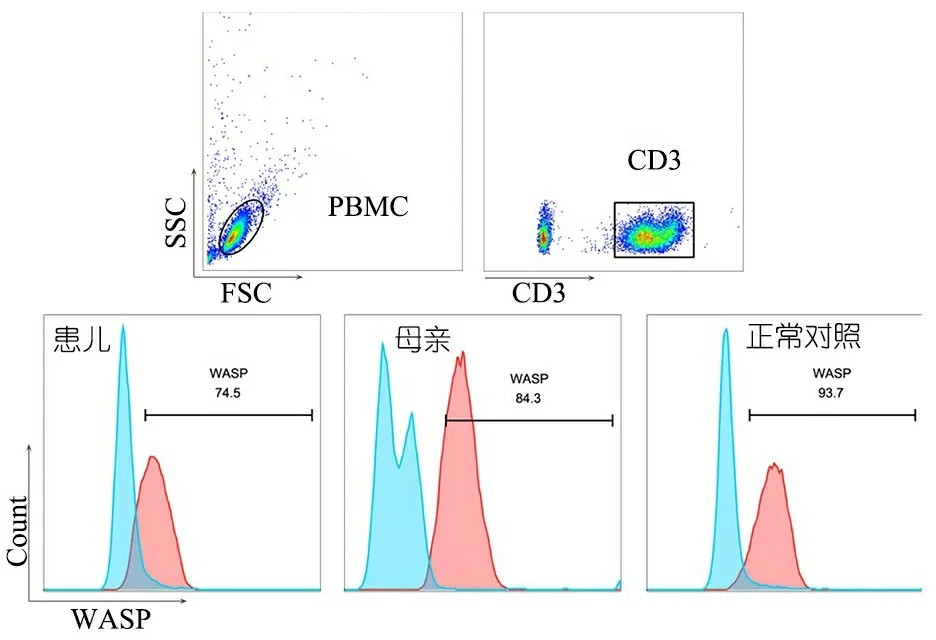
图4患儿及其母亲WASP表达
注 A图为设门方法,B图为患儿、其母亲和正常对照的WASP表达结果
治疗与随访:患儿入我院后感染症状不明显,ANC低,予粒细胞集落刺激因子(G-CSF)后ANC可从0.14×109·L-1上升至1.25×109·L-1,最高可升至3.32×109·L-1,随访1周后外周血ANC再次下降。随访至2018年9月,患儿病情稳定,发热和感染次数减少,外周血常规ANC(0.17~0.33)×109·L-1。
2 文献复习
检索PubMed、中国知网、万方和维普数据库,检索时间从建库至2018年9月20日。英文检索关键词为“X-linked neutropenia”,检出542篇;中文检索关键词为“X连锁中性粒细胞减少症”,检出0篇。筛选WAS基因突变导致的XLN病例的相关文献23篇,排除重复报道病例后,共2个家系(比利时和爱尔兰)和3例散发病例(刚果民主共和国和日本各1例,另1例国别不详),总计18例,共发现4个突变位点(L270P、S272P、I290T、I294T)[1-4,],2001年欧洲报道了第1个XLN家系,2018年日本报道了第1例亚洲XLN病例[5]。
对国外报道的本文18例及本文患儿(共19例)进行了临床资料总结和分析(表1)。患者均为男性,Sanger测序均发现WAS基因GBD结构域突变,ANC减少占84.2%(16/19),其中15例<0.5×109·L-1,单核细胞数量减少(13/18,72.2%),WBC数量降低(9/14,64.3%)。淋巴细胞亚群分析显示,NK细胞数量均明显降低(16/16),CD8+T细胞升高(7/16,43.7%)、CD4+/CD8+比值倒置(9/16,56.3%)。6例(54.5%)骨髓细胞学存在粒系成熟障碍,其中2例伴有染色体核型异常,2例伴红系/淋巴系发育异常,余2例仅有粒系成熟障碍。WAS基因功能获得性突变临床上主要表现为反复细菌感染(11/17, 64.7%),3例频率可达每年8~10次,1例表现为不明原因发热、2例无症状、3例ANC正常。患者基因突变常于成年后发现(10/14, 71.4%),此前仅2例患者在儿童期即发现基因异常[5],但其ANC处于正常水平,不能诊断为XLN。本文患儿是确诊年龄最小的XLN患者。
3 讨论
WAS基因突变常导致经典的Wiskott-Aldrich综合征(WAS),以PLT减少伴体积缩小、湿疹及反复感染为临床特征,病情常较危重,多因严重感染和出血而死亡[6]。WAS基因突变还可导致临床表型较轻的X-连锁血小板减少症(XLT)和X连锁中性粒细胞减少症(XLN)。相关疾病不同临床表型与WAS基因突变类型不同有关,无义突变和缺失突变导致WAS蛋白(WASP)表达缺失,引起严重的WAS综合征;错义突变导致WASP表达减少,引起XLT或者较轻的WAS综合征;WAS基因功能获得性突变则会引起XLN[7, 8]。XLN较其他两种疾病表型罕见。

表1 WAS基因功能获得性突变患者的临床特征
注 G-CSF:粒细胞集落刺激因子;MDS:骨髓增生异常综合征;NA:未检查;URTI:上呼吸道感染;AML:急性髓细胞白血病;ANC:中性粒细胞绝对计数;WBC:×109·L-1;N:中性粒细胞;M:单核细胞;L:淋巴细胞
WASP可调节细胞内肌动蛋白骨架生成,从而参与多种细胞生命过程,如细胞迁移、黏附聚集、分裂增殖等。WAS蛋白家族包括WASP、N-WASP、Scar/WAVE和Bee1,分别具有不同的组织特异性表达,其中WASP仅在血液系统表达。WASP在非激活状态下,C端的VCA结构域与GBD结构域结合,称为WASP的自身抑制。Cdc42是Rho家族成员,具有GTP酶活性,通过与GBD结构域结合激活WASP和N-WASP[9]。当VCA与GBD结合被Cdc42破坏后,WASP游离C端通过激活Arp2/3复合体[10],进而促进肌动蛋白聚合[11-13]。目前报告的XLN病例,其突变位点均位于WASP的GBD结构域,WASP自身抑制平衡被打破,造成Arp2/3复合体持续激活,导致肌动蛋白聚合异常,细胞极化异常,细胞凋亡增加,这在一定程度上解释了骨髓粒系成熟障碍[1-3, 14]。
XLN患者临床表现为ANC减少,部分患者有反复细菌感染,但与重型先天性中性粒细胞减少症(SCN)及经典WAS综合征患者相比,XLN患者感染相对较轻,导致了临床上容易漏诊及确诊年龄较晚[2]。研究报道,XLN患者唾液的中性粒细胞数量正常,XLN小鼠模型实验发现,由WAS功能获得性突变导致中性粒细胞过度激活,并向外周组织迁移增加[15]。这是否可以解释XLN患者虽然ANC数量低但感染并不严重的现象,有待进一步研究。
目前报道中对XLN患者的治疗主要参照SCN治疗方案,包括预防性应用抗生素和皮下注射G-CSF。G-CSF是快速升高ANC的最有效方法,可以明显改善患者预后和生存质量[16],但是长期使用存在并发血液系统恶变的风险,以急性髓细胞白血病(AML)和骨髓增生异常综合征(MDS)最为常见。国际重症慢性中性粒细胞减少症登记处(SCNIR)对长期大剂量使用G-CSF治疗SCN患者随访15年后发现,约22%转化为AML/MDS[17]。SCN患者长期使用G-CSF可造成CSF3R基因继发突变,该基因突变与血液系统恶变高度相关,在XLN病例中同样存在该现象[18]。2001年首次报道XLN家系的课题组[1]对该家系中5例患者长期随访,于2009年发现2例患者发生血液系统恶变[18],其中1例患者33岁起使用G-CSF 0.3 mg,每周3次,6年后发生AML,同时发现CSF3R基因g.2425T>G(p.Y729)突变;另1例患者63岁开始使用G-CSF 0.3 mg·d-1,持续皮下注射2年后发生MDS-RAEB,并发现CSF3R基因g.2390C>T (p.Q718)突变造成的CSF3R翻译提前终止。鉴于婴儿期因粒细胞减少导致脓毒血症而夭折的风险较大,而长期使用G-CSF又有引起CSF3R继发突变和导致血液系统恶变的风险,因而有研究者建议预防性使用抗生素作为基本治疗手段,并在综合考虑风险与收益的基础上,建议婴儿期使用G-CSF[3]。总之,对是否使用G-CSF,仍有争议,目前尚无对G-CSF治疗无反应或行骨髓移植治疗的XLN病例报道。
本例患儿是国内报道的第1例因WAS基因功能获得性突变导致XLN的病例,外周血ANC持续减少、骨髓粒细胞成熟障碍是重要的临床诊断线索,基因检测发现WAS基因突变可以明确诊断。与经典WAS综合征引起WASP表达缺失或低表达不同,XLN患者的WASP仍可表达,也是临床上容易忽视的重要因素。因此,对临床上怀疑先天性粒细胞减少症的患儿,需警惕WAS基因缺陷。
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
中国生殖健康(2020年2期)2021-01-18 02:51:26
英语学习(上半月)(2019年9期)2019-10-10 02:17:38
广州大学学报(自然科学版)(2019年1期)2019-05-07 01:33:26
小学生导刊(2018年13期)2018-06-29 03:49:00
米娜·女性大世界(2016年8期)2016-08-17 17:01:00
工业设计(2016年11期)2016-04-16 02:44:40
天津科技大学学报(2016年1期)2016-02-28 16:59:45
湖北师范大学学报(自然科学版)(2015年2期)2016-01-10 08:41:53
现代检验医学杂志(2015年2期)2015-02-06 02:01:01
