
基于密度泛函数理论的α-二亚胺镍配合物催化乙烯/降冰片烯共聚合的计算化学研究
2019-01-21 01:24:48崔咪咪薛小松侯彦辉刘宾元
天津工业大学学报 2018年6期
杨 敏,姜 湃,崔咪咪,薛小松,侯彦辉,刘宾元
(1.河北工业大学 化工学院,天津 300130;2.南开大学元素有机重点实验室,天津 300071;3.天津工业大学 材料科学与工程学院,天津 300387)
乙烯/降冰片烯共聚物被认为是最有前途的新型工业热塑性聚合物,具有高玻璃化温度、耐热性、良好的光学透明性、低介电常数以及优异的防潮性能等,因此,其可以广泛地应用于医用材料、电子元器件和光学材料[1-3].由于乙烯/降冰片烯(E-N)共聚物的优异性能,使用不同种类催化剂催化乙烯/降冰片烯共聚反应的研究已经引起了越来越多的关注.乙烯/降冰片烯加成共聚物可以通过各类催化剂进行催化合成,如茂金属催化剂[4-6]、前过渡非茂金属催化剂[7-9]以及后过渡非茂金属催化剂[10-12].另一方面,许多研究学者开始使用密度泛函理论(DFT)来研究乙烯/降冰片烯共聚反应[12-19]的反应机理.王永霞等[16]运用理论计算的方法发现在由半茂钛催化的乙、降共聚过程中,降冰片烯单体的引入将降低初始插入步骤的反应能垒,促进共聚反应的发生.理论计算也被应用于探究金属钒作为活性中心催化乙烯均聚、乙烯/降冰片烯共聚的反应机理[17].Tang等[18]运用密度泛函理论探究半茂钛金属配合物催化的乙烯/降冰片烯共聚反应机理,取得显著成果.Kim等[19]运用密度泛函理论探究乙烯和降冰片烯在插入金属活性中心过程中的竞争关系,发现降冰片烯单体上的环戊基展现出固定氢键的作用,加强降冰片烯单体插入活性中心的能力.
α-二亚胺镍催化剂体系被广泛地应用于乙烯均聚[20-24]、降冰片烯均聚[25-28]以及乙烯、降冰片烯嵌段共聚[29]反应中.然而,使用该催化体系催化乙烯和降冰片烯无规共聚反应以及运用密度泛函理论来探究该无规共聚过程的反应机理的研究未见报道.本课题组将N,N′-二(2,6-二异丙基)苊二亚胺溴化镍催化剂应用于乙烯、降冰片烯均聚以及无规共聚反应,但研究结果发现该催化剂仅能催化均聚反应,而无法催化乙烯/降冰片烯无规共聚反应.为了探寻造成这一实验结果的原因,本文使用密度泛函理论(DFT)方法模拟共聚过程中可能出现的所有反应结构,计算相应结构的势能,并对乙烯和降冰片烯的引发反应、再插入反应以及乙烯的线性链增长和β-氢转移反应分别进行能量对比.
1 实验部分
1.1 实验材料
所有对水、氧敏感的操作均在氩气气氛下的手套箱抑或是Schlenk线性管路中进行.N,N′-二(2,6-二异丙基)苊二亚胺溴化镍参照文献[30-31]进行合成,其平面结构如图1所示;活化剂一氯二乙基铝(AlEt2Cl,1.0 mol/L)、甲基铝氧烷(MAO,1.5 mol/L),Albemarle公司产品;乙烯气体,天津赛美特特种气体有限公司产品;甲苯在使用前需经过钠/二苯甲酮蒸馏干燥操作;氯苯在使用前经过氢化钙干燥处理.所有其他的化学品均直接购买.

图1 N,N′-二(2,6-二异丙基)苊二亚胺溴化镍配合物的平面结构Fig.1 Planar structure of bis[N,N′-(2,6-diisopropylphenyl)imino] acenaphthene dibromonickel
1.2 乙烯、降冰片烯的均聚和无规共聚反应
聚合反应在100 mL具有机械搅拌功能的不锈钢高压釜中进行.在进行聚合实验之前,高压釜需在150℃下抽真空2 h,然后在氩气环境下冷却至室温,最后用干燥的氩气置换2次,乙烯置换1次.实验时,在指定温度下,依次注入溶剂甲苯、定量催化剂和助催化剂,充分混合后充入乙烯气体达到指定压力.反应结束时,使用10%酸化乙醇终止聚合反应.聚合产物使用蒸馏水和无水乙醇反复清洗3遍,置于60℃下的真空烘箱干燥24 h.
1.3 计算方法
对于聚合反应,反应过程的势能和结构计算充分参照 Brookhart-Green[30]和 Cossée-Arlman[32]机理.本文所有的反应物、中间体、过渡态、产物的结构优化和能量计算均使用密度泛函数理论方法.反应过程中的所有吉布斯自由能的数值采用(SMD)B3LYP/[6-31G(d)+Lanl2dz]//(SMD)M06/[6-311++G(d,p)+SDD]水平进行计算.结构优化、振动频率以及热力学校正使用B3LYP方法,并对金属镍使用Lanl2dz赝势基组,而其余所有原子使用6-31G(d)基组[33-34].甲苯溶剂的影响采用SMD溶剂化模型进行计算[35].所有结构优化的结构均采用频率计算的方法进行过渡态抑或是稳定结构的验证.
为了获得更加精确的计算值,在(SMD)M06/[6-311++G(d,p)+SDD]水平下对已经经过(SMD)B3LYP/[6-31G(d)+Lanl2dz]水平优化过的稳定结构进行单点能计算.单点能计算中使用M06方法,对金属镍采用SDD赝势基组,对其余所有原子采用6-311++G(d,p)基组.所有结构使用CYL软件进行观测.所有计算均使用Guassian09软件进行,所有计算能量单位均使用kcal/mol(1 kcal=4.186 kJ).
2 结果与讨论
2.1 α-二亚胺镍配合物的催化活性
N,N′-二(2,6-二异丙基)苊二亚胺溴化镍作为主催化剂A(catalyst A),甲基铝氧烷或是一氯二乙基铝做助催化剂,催化乙烯均聚、降冰片烯均聚和乙烯/降冰片烯无规共聚反应,研究不同催化条件包括反应温度、反应时间、反应压力以及铝镍比对催化剂活性所产生的影响,结果如表1所示.
由表1可以看出,在乙烯均聚反应中催化剂活性达到2.91×106g/(mol·h),在降冰片烯均聚反应中催化活性可达0.45×105g/(mol·h).然而在各种反应条件下,乙烯和降冰片烯无规共聚反应均无聚合产物.因此,取共聚反应后的反应液进行气质联用分析,谱图显示反应液中的主要成分是反应溶剂和反应单体.这个结果意味着该催化体系催化乙烯/降冰片烯无规共聚反应无法产生共聚产物.那么,为什么该催化体系能催化乙烯和降冰片烯均聚反应,而无法催化两者的无规共聚反应?为了探究这些原因,本文采用密度泛函数理论计算方法,模拟共聚过程所有可能发生的反应结构并计算相应结构的能量.
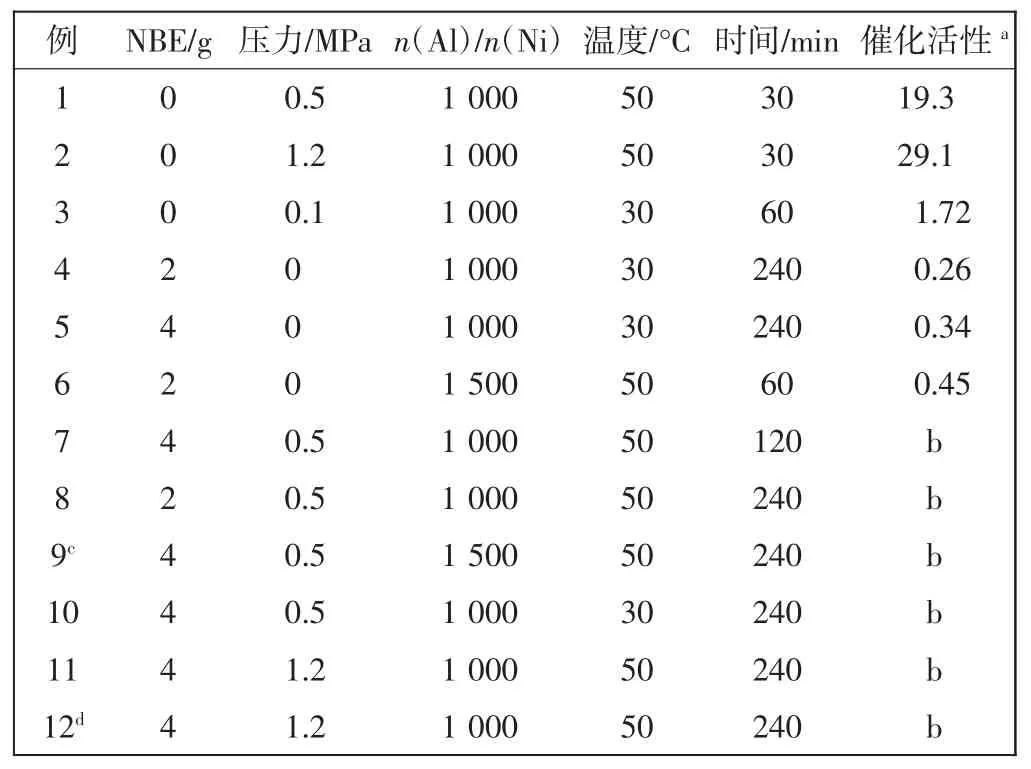
表1 催化剂A催化乙烯、降冰片烯均聚和共聚Tab.1 Homo-and copolymerization of norbornene and ethylene catalyzed with catalyst A
2.2 乙烯和降冰片烯插入Ni-CH3键过程对比
基于实验结果,本文运用DFT理论研究降冰片烯和乙烯单体分别插入阳离子金属活性中心的过程,模拟插入过程可能产生的结构并计算其对应结构的势能,如图2—图4所示.

图2 对于乙烯引发,阳离子引发结构1、配合物结构2/2a、过渡态结构3、产物结构4的分子结构Fig.2 For ethylene initiation,top view of molecular geometries of cation complex 1,π-complex 2,π-complex 2a,transition state 3 and product 4

图3 对于降冰片烯引发,配合物结构5/5a、过渡态结构6/6a、产物结构7的分子结构Fig.3 For norbornene initiation,top view of molecular geometries of π-complex 5,π-complex 5a,transition state 6,transition state 6a and product 7

图4 乙烯和降冰片烯插入阳离子活性中心势能图Fig.4 Calculated potential energy diagram for norbornene and ethylene insertion into Ni-CH3bond of[Ni(CH3)]+
根据文献[30,36]的研究结果,可假设初始引发反应从阳离子复合物结构1开始,复合物结构2具有T型平面(Cs)结构且处于能量最低态,如图2所示.乙烯插入阳离子活性中心的第一步是乙烯单体与复合物1进行配位.在配位过程将产生2种具有Cs对称的π配位结构(2和2a),2种结构区别在于是平行或垂直于Ni(N=C—C=N)平面.从图4所示势能图可以发现,复合物2的势能比结构2a低10 kcal/mol,因此复合物结构2是更加稳定的结构.在反应过程中,结构2比结构2a更容易形成.形成配合物结构2后,经过渡态结构3(TS3)生成反应物结构4.Hessian分析证实结构3具有269.27i的虚频,是乙烯插入阳离子活性中心过程的过渡态结构.基于图4中的势能基线,乙烯单体插入Ni—CH3键过程的总能垒为-4.7 kcal/mol.
降冰片烯插入阳离子活性中心的第一步是降冰片烯单体与复合物1配位形成配合物结构5,然后通过过渡结构6生成产物结构7,如图3所示.对于降冰片烯单体与阳离子催化活性种的配位过程存在endo和exo 2种构型,因此,在降冰片烯插入阳离子活性中心的过程中也存在2种可能的反应方式.通过单点能计算,配合物中间体结构5a的能量为-7.8 kcal/mol,处于势能浅井,结构稳定性远不如势能更低的中间体结构5,如图4所示.因此,中间体结构5是更稳定的结构,并将优于结构5a率先形成.对于过渡态结构6(TS6)和 6a(TS6a),它们分别具有 237.92i和 238.16i的虚频,但它们具有相同的势能(-7.4 kcal/mol).综上所述,降冰片烯插入阳离子活性中心反应的总能垒为-7.4 kcal/mol.
对比降冰片烯单体和乙烯单体插入阳离子活性中心反应的总能垒,发现降冰片烯单体在插入Ni-CH3键反应过程中具有更低的反应能垒,将更容易发生.因此,从理论上说明共聚过程中降冰片烯单体更占优势,将比乙烯单体优先进行插入反应,此时体系中主要存在结构7.
2.3 乙烯和降冰片烯插入Ni—NBE结构过程比较
根据上述计算结果,将结构7作为再插入反应过程的初始结构,模拟再插入过程可能产生的结构并计算其对应结构的势能,结果如图5—图7所示.
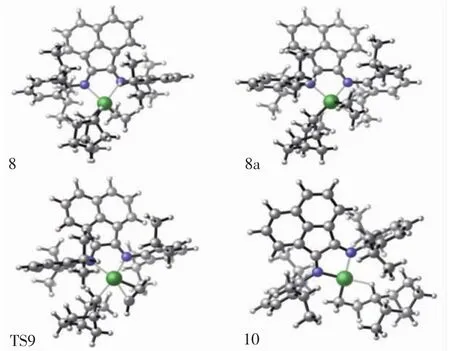
图5 配合物结构8/8a、过渡态结构9,产物结构10的分子结构Fig.5 Top view of molecular geometries of π-complex 8,π-complex 8a,transition state 9 and product 10
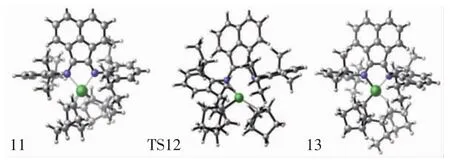
图6 配合物结构11、过渡态结构9、产物结构13的分子结构Fig.6 Top view of molecular geometries of complex 11,transition state 9 and product 13

图7 降冰片烯和乙烯再插入反应势能图Fig.7 Calculated potential energy diagram for norbornene and ethylene insertion into complex 7
对于乙烯单体的再插入反应,首先是乙烯单体对结构7进行配位,由于结构7的空间不对称性,乙烯单体可从结构7后侧或是前侧进行配位,因此,本文模拟2种进攻方式的结构,并计算其能量.经过单点能计算出图5中配合物结构8a(单体后侧进攻)的势能为4.51 kcal/mol,配合物结构8的势能为-2.26 kcal/mol.对比2种进攻方式的势能发现,中间体结构8a处于势能浅井,比8具有更高的势能,如图7所示.因此,中间体结构8更为稳定,将优于结构8a率先形成.随着中间体结构8的形成,乙烯再插入反应将通过过渡态结构9(TS9)生成最终产物结构10.观察乙烯单体插入Ni—CH3键反应过程和插入Ni—NBE键反应过程,发现初始插入反应的过渡态3(TS3)与再插入反应的过渡态9(TS9)在结构上非常相似,如图2和图5所示.乙烯再插入反应过程的过渡态势能为9.98 kcal/mol,比乙烯初始插入反应的过渡态势能高2.58 kcal/mol.
对于降冰片烯插入Ni-NBE键反应,首先是降冰片烯单体与结构7进行配位,形成配位中间体结构11,然后通过过渡状态12(TS12)获得产物结构13,如图6所示.与此同时可发现,降冰片烯再插入过程的过渡态结构12(TS12,图6)和初始插入反应的过渡态结构6(TS6,图3)也是非常相似的.
单点能计算结果表明,降冰片烯再插入过程的中间体结构11的势能是12.12 kcal/mol,而乙烯再插入过程的中间体结构8的势能是-2.26 kcal/mol,这表明乙烯比降冰片烯更容易与Ni—NBE结构进行配位.降冰片烯再插入反应的总能垒为18.49 kcal/mol,也远高于乙烯单体再插入反应的总能垒(9.98 kcal/mol).因此,在体系中乙烯的再插入反应将比降冰片烯的再插入反应更容易发生.相关研究[16]也报道称,由于降冰片烯的自身空间位阻,其连续插入反应是很难发生的.因此,单体再插入Ni—NBE键反应过程中,乙烯单体比降冰片烯更容易完成插入反应,所以结构10将是此阶段主要产物结构.
2.4 乙烯的β-氢转移反应和线性链增长反应的对比
乙烯线性链增长反应和β-氢转移反应的理论计算研究[37-38]结果如图8—图10所示.

图8 配合物结构14、过渡态结构15、产物结构16的分子结构示意图Fig.8 Top view of molecular geometries of π-complex 14,transition state 15 and product 16
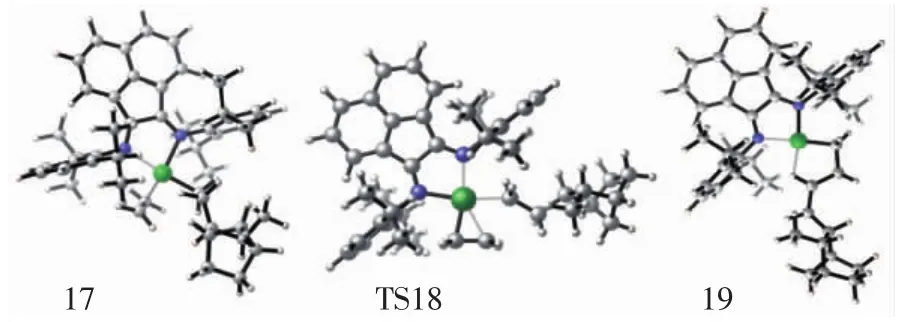
图9 配合物结构17、过渡态结构18、产物结构19的分子结构Fig.9 Top view of molecular geometries of π-complex 17,transition state 18 and product 19

图10 乙烯线性增长反应和β氢转移反应的势能图Fig.10 Calculated potential energy diagram for β-hydride transfer reaction between linear propagation
对于乙烯β-氢转移反应,其过渡态是非常难以寻找的.但根据文献[38]的研究结果,构建出中间体结构15,该结构15与β-氢转移反应过程的过渡态结构在能量和空间构型上是非常相似的.因此,本文把中间体结构15作为β-氢转移反应的过渡态,如图8所示.该反应从结构10开始,经历结构14(β-agostic),然后通过过渡态结构 15(TS15)形成结构 16(hydride-olefin).中间体结构14的势能是-9.35 kcal/mol,β-氢转移反应的总能垒是1.17 kcal/mol,如图10所示.
对于乙烯线性链增长过程,乙烯单体首先与结构10配位形成π复合物结构17,该配位结构与乙烯插入Ni—CH3过程中间体结构2也是相似的.随着π复合物17的形成,链增长反应通过过渡态结构18形成产物结构19,如图9所示.通过单点能计算表明,π配合物结构17的势能是-8.6 kcal/mol,乙烯链增长过程的总能垒为6.0 kcal/mol,如图10所示.
比较上述2个反应过程,发现β-氢转移反应中的络合过程比乙烯线性链增长的配位过程更容易发生,且乙烯线性链增长反应过程的总能垒要远高于β-氢转移反应过程的总能垒.因此,在该阶段乙烯的β-氢转移反应将比乙烯线性链增长反应更容易发生.
2.5 乙烯/降冰片烯无规共聚过程分析
对于本文采用催化剂形成的阳离子活性中心,通过计算乙烯/降冰片烯无规共聚过程中所有可能发生的主要反应,研究发现降冰片烯单体将比乙烯更容易发生插入反应,而在随后的再插入过程中,由于降冰片烯单体自身的大空间位阻的影响,其连续插入是十分困难的,因此,再插入过程中乙烯将更容易发生反应.随后,在乙烯不断增长过程中,β-氢转移反应将导致链段断开,且转移过程比线性链增长反应更容易发生.观察β-氢转移反应的终止结构与阳离子活性中心结构,发现两者具有的结构是十分相似的,因此,单体可以再次进行插入反应.综上所述,降冰片烯插入Ni-CH3键的引发反应、乙烯的再插入反应和增长链的β-氢转移反应将构成循环体系,如图11所示.该循环体系将导致乙烯/降冰片烯无规共聚反应只产生短链分子而鲜有长链聚合物产生,从理论上解释了催化剂A催化乙烯和降冰片烯无规共聚无产物的原因.
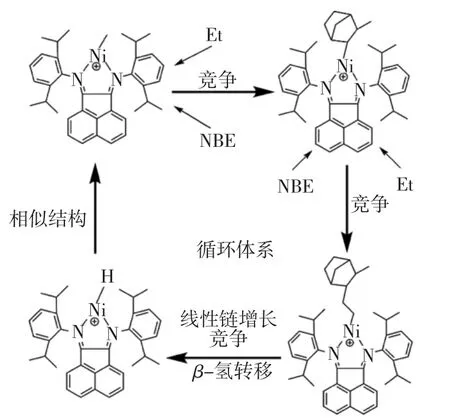
图11 乙、降无规共聚过程的每步反应的平面结构Fig.11 Planar structure of each step in ethylene and norbornene copolymerization
3 结论
本文使用密度泛函数理论方法对N,N′-二(2,6-二异丙基)苊二亚胺溴化镍催化的乙烯/降冰片烯无规共聚的反应过程进行研究,发现:在共聚反应中,降冰片烯单体将降低初始插入反应过程的能垒,优先乙烯插入阳离子活性中心,而乙烯将更容易发生随后的再插入反应;但随着链段增长,β-氢转移反应将打断链段的增长,导致该体系始终无法生成长链结构.通过理论计算,解释了使用含有苊醌配体的二亚胺镍化合物催化乙烯和降冰片烯无规共聚反应无聚合产物生成的原因.
猜你喜欢
中学生数理化·八年级物理人教版(2023年6期)2023-05-25 11:59:48
——《势能》
文化纵横(2022年3期)2022-09-07 11:43:18
北京航空航天大学学报(2022年5期)2022-06-06 09:27:18
中学生数理化·八年级物理人教版(2022年6期)2022-06-05 06:55:40
中学生数理化·八年级物理人教版(2021年6期)2021-11-22 07:49:52
大学化学(2021年8期)2021-09-26 10:51:16
广州中医药大学学报(2021年6期)2021-05-23 12:32:44
中成药(2019年12期)2020-01-04 02:03:00
中国医药指南(2019年10期)2019-01-07 07:21:20
电脑知识与技术(2018年3期)2018-03-21 09:27:04
