
聚二甲基硅氧烷/纳米纤维素复合膜的制备及性能分析
2019-01-10 08:03:40陈楚楚王怡仁卜香婷罗德丹李大纲
纤维素科学与技术 2018年4期
陈楚楚, 王怡仁, 卜香婷, 罗德丹, 李大纲*
(南京林业大学,材料科学与工程学院,江苏 南京 210037)
聚二甲基硅氧烷(polydimethylsiloxane, PDMS)是一种高分子有机硅化合物[1],因其具有良好的热稳定性、柔韧性及生物相容性等优点[2],使其作为基底材料广泛应用于生物微纳传感器[3]、组织工程生物材料等领域[4]。纳米纤维素(cellulose nanofibers,CNF)具有高强度、高比表面积、生物可再生性等优点[5],其中纤维素结晶区的杨氏模量可达138 GPa, 拉伸强度约3 GPa,是一种理想的纳米尺度增强相[6],广泛用于提高聚合物的力学性能[7]。
将纳米纤维与聚合物混合,制备高强度复合材料是提高聚合物力学强度的常见方法之一[8-10]。Sang等[10]通过混合法制备了生物基聚氨酯(BPU)/ 纤维素纳米晶须复合材料,与纯BPU相比,改性纤维素纳米晶须(M-CNW)的添加(纳米纤维素质量分数分别为0.5%、1%、5%)显著增强了BPU复合材料的拉伸强度。Liu等[11]使用混合法制备了天然橡胶(NR)/ 甲壳素纳米晶(CNCs)复合材料(甲壳素纳米晶质量分数1%到10%),CNCs在NR中的均匀分散有效地了提高橡胶的机械性能和热稳定性。与纯聚合物相比,将纳米纤维素通过混合法与聚合物结合,有助于提高复合材料的力学性能,然而随着纳米纤维素在聚合物中质量分数的提高,其复合材料强度与弹性模量呈下降趋势;这是由于纳米纤维素比表面积较大,在聚合物基材内难于分散且易发生团聚[9],当外力作用时易发生应力集中,从而导致其力学性能的下降。
故本次研究采用浸渍法,首先通过溶剂置换法将纳米纤维素薄膜内部水分置换为正己烷,再浸泡至不同浓度PDMS预聚体溶液中,经加热固化后制得具有不同PDMS质量分数的PDMS/CNF复合材料,进而对其力学性能、微观形态、晶型结构等进行表征分析。与传统混合法相比,浸渍法能够显著提高纳米纤维增强相在聚合物基材中的分散浓度,有助于进一步改善复合材料的力学性能。
1 实验
1.1 材料及仪器
竹粉,目数60~80,自制;道康宁184 PDMS预聚物液体硅橡胶购自美国道康宁公司;乙酸、正己烷、亚氯酸钠及氢氧化钾(KOH)等试剂均为分析纯,购于南京化学试剂有限公司。使用仪器包括:FE-SEM场发射扫描电子显微镜(JSM-6700F,JEOL Ltd,Japan),Nicolet iS10型傅里叶红外光谱仪,X-射线衍射仪(UltraX 18HF,Rigaku,Japan)以及万能力学实验机(Model3365;Instron Corp.,Canton,MA)。
1.2 实验步骤及方法
1.2.1纤维素的提纯处理
称取30 g竹粉置于烧杯中,加1 700 g去离子水,搅拌后静置10小时。取16 g 纯度为80%的亚氯酸钠及2.4 mL乙酸配置酸溶液,倒入浸泡竹粉的烧杯中。将烧杯置于水浴锅中,在80℃下加热搅拌1小时,取出冷却至室温,用真空抽滤器冲洗过滤直至溶液呈中性,此步骤重复四次。取酸处理样品置于烧杯中,加入6%(wt)KOH溶液700 mL,靜置10小时后。放入水浴锅中,在90℃下加热搅拌2小时,取出用真空抽滤器冲洗至溶液呈中性。将所得纯化纤维素样品置入烧杯中,密封待测。
1.2.2纳米纤维素的制备
取上述酸碱提纯处理过后的纤维素样品,加入蒸馏水配制成浓度为 0.8%~1%(wt)的悬浮液,并将其倒入研磨机中,调节研磨机转速为1 500 r/min,磨盘与磨盘间间距为-2.5(-0.25 mm),进行一次研磨处理,收集最终所得的浓浆样品为纳米纤维素[12]。取部分样品于100℃烘箱烘干,称取前后重量,计算出该样品的浓度并记录。
1.2.3纳米纤维素膜的制备
取含纤维素0.05 g的悬浮液,加入适量蒸馏水稀释至0.1%(wt),用超声波细胞破碎仪进行超声处理20分钟。将得到的混合悬浮液倒入抽滤装置进行真空抽滤,制得纳米纤维素薄膜,如图1所示。
1.2.4聚二甲基硅氧烷/纳米纤维素薄膜的制备
由于PDMS不溶于水,溶于正己烷等有机溶剂,故采用“水-乙醇-正己烷”逐级溶剂置换法,将纳米纤维素薄膜依次浸泡至无水乙醇、正己烷中进行溶剂置换,置换6次,每次4小时,直至薄膜中水分完全置换为正己烷溶液。取 PDMS预聚体溶液置于烧杯中,加入正己烷进行稀释,配置成不同浓度的 PDMS预聚体溶液(浓度分别为1%、3%、5%以及10%)。将溶剂置换处理后的CNF薄膜放入PDMS预聚物溶液中浸泡12小时后,滴加固化剂(固化剂与PDMS预聚物的比例为1∶10),混合均匀后静置30分钟。30分钟后取出CNF薄膜,刮去表面多余溶液后放入60℃烘箱中加热进行预烘干,待薄膜表面溶剂挥发后置于两片聚四氟乙烯板之间,放入60℃烘箱中烘干2小时,使PDMS完全固化,所得样品即为PDMS/CNF复合薄膜,如图1所示。将制备所得复合薄膜用PDMS/CNF-n表示,n为浸泡时PDMS预聚体溶液的质量分数(n=1、3、5、10)。
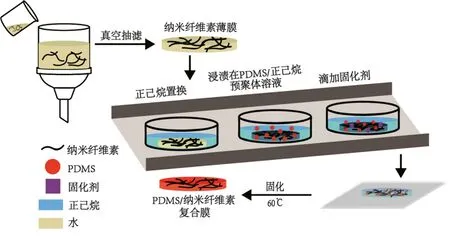
图1 PDMS/CNF复合薄膜制备流程
1.3 测试分析方法
通过FE-SEM场发射扫描电子显微镜(JSM-6700F,JEOL Ltd,Japan)表征PDMS/CNF复合膜的形貌特征。将待测样品烘干后置于导电碳膜上,采用自动喷金仪对样品进行喷金处理,扫描电压为 1.5 kV,电流为 10 µA。
使用Nicolet iS10型傅里叶红外光谱仪,在 Smart iTR diamond ATR 模式下对PDMS/CNF复合膜的化学结构进行表征分析。测试扫描速度为0.2 cm/s,波数扫描范围为650~4000 cm-1,测试前对样品进行干燥处理。
利用 X-射线衍射仪(UltraX 18HF,Rigaku,Japan)分析CNF、PDMS及其复合材料的结晶性能。测试采用铜靶,管电压40 kV,管电流为300 mA,扫描角度范围为2θ=5º~40º,扫描速度为 5º/min。
将待测样品膜裁切成宽度为5 mm,长度为35 mm。测量试样的厚度、宽度并记录,将试样夹在夹具间,保证跨度在20~30 mm之间。设定万能力学实验机(Model3365;Instron Corp.,Canton,MA)的载荷为1 kN,加载速度为10 mm/min。
2 结果与讨论
2.1 微观形貌分析
将CNF薄膜浸泡入不同浓度的PDMS溶液中,固化后制得PDMS/CNF复合膜。经公式1计算,随PDMS浸泡浓度的提高,制得复合膜中PDMS的质量分数逐渐增加。PDMS/CNF-1、3、5、10中PDMS的质量分数为 18%、25%、35%、48%。PDMS质量分数(W)由 PDMS/CNF复合薄膜的质量(M1)与CNF薄膜的质量(M2)求得,如公式(1)所示。

利用FE-SEM观察样品的微观形貌特征,进一步了解PDMS与CNF间的分布。图2a所示为PDMS,其表面较光滑且平整。图2b所示CNF呈现三维纳米孔洞结构,其中CNF纤维直径约30~50 nm。图2c、2d所示为PDMS/CNF-1分别放大10 000倍与100 000倍后的表面形貌。从图中可看出,PDMS胶状物质填充在纤维网状结构的孔洞之中,使其表面呈现较为平整的形态;进一步放大可以清晰观察到被PDMS包裹后的纤维结构。图2e、2f所示分别为PDMS/CNF-10放大20 000与100 000倍后的表面形态。与PDMS/CNF-1相比,提高PDMS质量分数后,其复合膜(PDMS/CNF-10)中,PDMS填充更为密集,纤维网状结构中可明显观察到PDMS胶状物质聚集体。由此可知,采用浸渍法所制得PDMS/CNF复合膜中,CNF以三维纳米网络形态嵌于PDMS中。随着PDMS浸泡溶液浓度的增大,PDMS聚集程度增加,更多地包裹在纤维表面及纤维网络的孔洞中。

图2 CNF、PDMS、PDMS/CNF扫描电镜图片
2.2 X-射线衍射(XRD)结晶结构分析
本研究利用 X-射线衍射分析制备过程中样品的晶型结构变化。纤维素的特征晶面(101、10ī、002、040)所对应角度分别为 14.98º、16.7º、22.8º、34.49º[13],图 3所示分别为 PDMS、CNF、PDMS/CNF-1、PDMS/CNF-10 X-射线衍射图。由图可以观察到PDMS/CNF-1、PDMS/CNF-10与CNF的特征峰型基本一致,均保留CNF原有的特征晶型结构。其中,在 14º~17.5º出现对应纤维素晶体的(101)和(10ī)晶面,在22.5º附近出现对应于纤维素晶体的(002)衍射晶面。这表明在整个复合处理过程中,纤维素其自身的晶型结构并没有改变。

图 3 PDMS、CNF、PDMS/CNF-1、PDMS/CNF-10 X-射线衍射图谱
2.3 FT-IR 光谱分析
图 4为CNF、PDMS、PDMS/CNF-1、PDMS/CNF-3、PDMS/CNF-10 FT-IR光谱图。如图 4所示,在PDMS/CNF复合薄膜中,波数3343 cm-1(O-H的伸缩振动峰)[14]、1413 cm-(1CH2的剪式振动峰)、1162 cm-1、896 cm-1(C-O-C不对称伸缩振动峰)为纤维素I的特征峰,1640 cm-1处的特征峰为纤维素分子中O-H的弯曲伸缩振动峰[15]。波数786 cm-1、1258 cm-1附近的吸收峰为二甲基硅氧烷的 CH3的伸缩振动,1012 cm-1附近的双峰属于Si-O-Si的不对称伸缩振动,2962 cm-1附近的是Si(CH3)2中CH3的伸缩振动吸收峰,为聚二甲基硅氧烷的特征峰[16]。比较PDMS与CNF复合前后样品的曲线,复合膜中各物质的特征峰均存在;随着PDMS质量分数的提高,由于PDMS大量充填于CNF三维纳米网络结构中或包裹于纤维表面使得复合薄膜中CNF特征峰强度逐渐减弱。
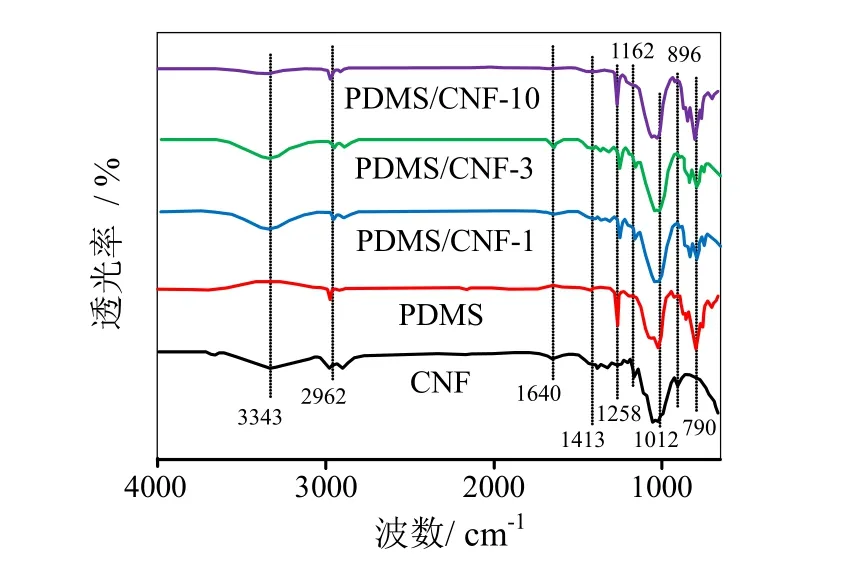
图 4 CNF、PDMS、PDMS/CNF-1、PDMS/CNF-3、PDMS/CNF-10 FT-IR光谱图
2.4 力学性能分析
表1为拉伸测试数据表。由表1可知,PDMS在复合薄膜中的质量分数随浸泡比例(1%、3%、5%、10%)上升而逐渐增大。其中复合薄膜的断裂伸长率随PDMS的增加而逐渐增大,拉伸强度逐渐下降。这是由于PDMS与CNF复合之后,CNF作为增强相极大的提升了复合薄膜的强度,PDMS作为填充相显著改善了复合薄膜的柔韧性。与传统混合法相比,浸渍法能够进一步提高CNF在PDMS中的分散浓度,当PDMS浓度为1%(wt)时,PDMS/CNF中CNF质量分数可达82%(wt),其拉伸强度较纯PDMS,由1 MPa提高至33 MPa。这是由于在拉伸过程中,复合薄膜受外力产生形变时,CNF纳米网络可将外力均匀分散,极大的提高了复合薄膜的强韧性。与纯CNF薄膜相比,PDMS/CNF-1复合膜的断裂伸长率由2.2%上升到5.4%,PDMS/CNF-10复合膜的断裂伸长率达到10.7%,其断裂伸长率为纯CNF薄膜的5倍。当PDMS质量分数进一步提高至48%,制备所得PDMS/CNF-10复合薄膜的拉伸强度较CNF薄膜由41.72 MPa降低至22.15 MPa,这是由于浸渍法复合PDMS和CNF时,CNF三维纳米网络中大量填充/聚集了胶状PDMS,造成复合薄膜拉伸强度的下降。
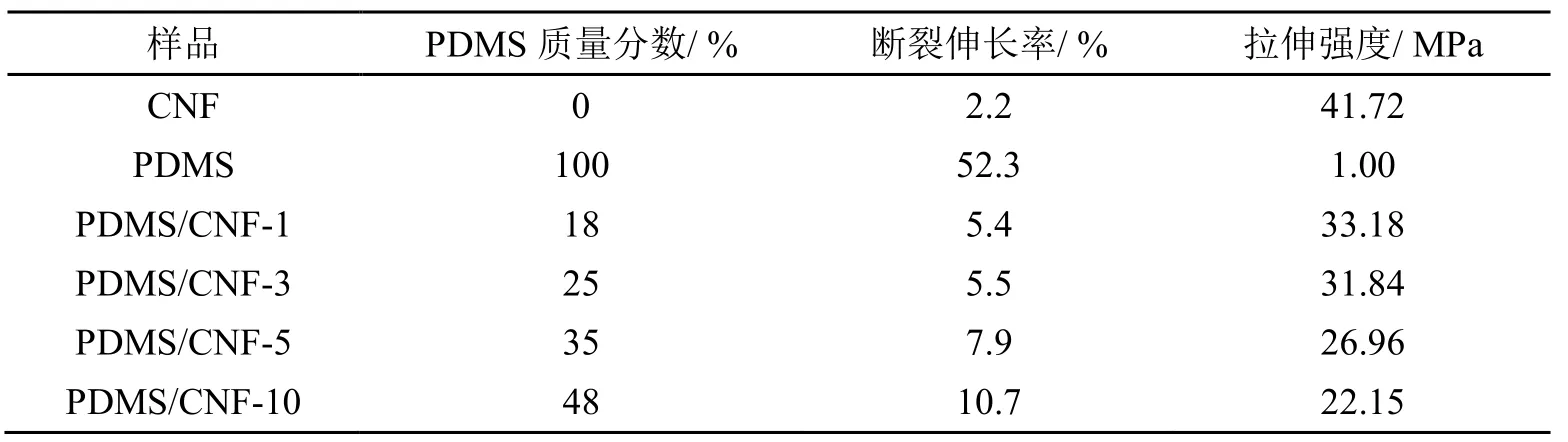
表1 拉伸测试数据表
3 结论
通过浸渍法制备 PDMS/CNF复合薄膜,并对其力学性能、形貌特征、晶形结构、化学组成等进行表征分析,结论如下:
1)FE-SEM表征结果表明,采用浸渍法制备所得PDMS/CNF复合薄膜,PDMS能够均匀填充于CNF三维纳米网络的多孔结构中,且固化后仍能保持其原有的纳米纤维形态。
2)XRD与FT-IR测试结果表明,复合前后样品的晶形结构(CNF)与化学组分并未发生变化。
3)拉伸力学测试结果表明,与传统混合法相比,浸渍法可以显著提高CNF在PDMS中的分散浓度,当PDMS浸渍浓度为1%(wt)时,制备所得PDMS/CNF复合膜中CNF质量分数可达82%(wt),其拉伸强度较纯PDMS,由1 MPa提高至33 MPa。
猜你喜欢
石油炼制与化工(2022年2期)2022-02-15 11:42:26
纺织科技进展(2021年3期)2021-06-09 08:07:14
应用化工(2021年4期)2021-05-20 09:43:36
陶瓷学报(2021年1期)2021-04-13 01:33:02
化工管理(2020年26期)2020-10-09 10:05:16
山东化工(2019年2期)2019-02-21 09:29:32
西南国防医药(2016年6期)2016-12-01 06:00:58
中国塑料(2016年1期)2016-05-17 06:13:10
中国塑料(2015年3期)2015-11-27 03:42:15
中国塑料(2014年10期)2014-10-17 03:06:19
