
氯化铵改性UF/E自修复微胶囊的性能表征
2018-12-27 13:15:02王信刚谢昱昊
建筑材料学报 2018年6期
王信刚, 徐 伟, 谢昱昊, 夏 龙
(南昌大学 建筑工程学院, 江西 南昌 330031)
自2001年White等利用微胶囊技术将修复材料包裹于脲醛树脂中,制备出具有核壳结构的自修复微胶囊之后,采用脲醛树脂作为壁材来制备自修复微胶囊就成了研究热点[1-3].氯化铵作为脲醛树脂微胶囊改性剂,可用于改变微胶囊壁材特性[4-6].Fan等[7-8]、Niu等[9]研究发现掺入氯化铵会导致反应过程中体系的pH值下降,加速自修复微胶囊的固化,增加脲醛树脂纳米颗粒在微胶囊表面上的沉积.Li等[10]研究发现掺入氯化铵能够降低自修复微胶囊中的残余甲醛含量.Tang等[11]研究发现相比于未掺入氯化铵,掺入氯化铵的微胶囊具有更好的耐热性和密封性能.上述研究基本都是通过简单掺入氯化铵来制备出自修复微胶囊,而对于氯化铵掺量对脲醛树脂微胶囊颗粒特征、囊化指标等影响的研究鲜有报道.
本文以环氧树脂E-51为芯材,脲醛树脂为壁材,通过两步原位聚合法制备UF/E自修复微胶囊,研究氯化铵用量(质量分数,本文所涉及的用量、含量、比值等均为质量分数或质量比)对自修复微胶囊微观形貌、粒径分布、产率与芯材含量的影响,同时采用傅里叶变换红外光谱(FTIR),热重分析(TGA)和环境扫描电镜(ESEM)来表征其化学结构和热稳定性.
1 试验
1.1 原材料
环氧树脂E-51(E),工业纯,广东穗欣化工有限公司产;质量分数为37%的甲醛水溶液、尿素、三乙醇胺、氯化铵、十二烷基苯磺酸钠SDBS和间苯二酚,均为分析纯,国药集团化学试剂有限公司产.
1.2 自修复微胶囊的制备
将尿素和质量分数为37%的甲醛水溶液按质量比1∶2混合,待尿素全部溶解后,用三乙醇胺调节溶液pH值至8.0~9.0,在70℃、搅拌速率450r/min下反应1h,得到脲醛树脂预聚体(UF prepolymer).芯材环氧树脂E-51在50℃,具有一定浓度的SDBS溶液中乳化40min,搅拌速率为1300r/min.将脲醛树脂预聚体加入乳化好的芯材乳液中(m(E)∶m(UF prepolymer)=1∶1),再用氯化铵和氢氧化钠溶液调节其pH值至3.0左右,在50℃、搅拌速率900r/min下反应1h.最后加入适量的间苯二酚,再升温至60℃,以900r/min的搅拌速率反应1.5h.经沉淀、过滤,烘干和分筛后得到UF/E自修复微胶囊.
1.3 试验方法
采用Quanta200F型环境扫描电子显微镜来表征自修复微胶囊的微观形貌;采用ZBSX-92A型震击式标准振筛机测定自修复微胶囊的粒径分布;采用Nicolet5700型傅里叶变换红外光谱仪对自修复微胶囊进行化学结构分析;采用TGA4000型热失重分析仪对自修复微胶囊进行热稳定性分析.
产率测定:在反应结束后去除多余的壁材、未反应的壁材及未包覆的芯材后,所得到的微胶囊干燥试样质量为m1,合成中投入反应体系的所有原料质量为m2(100g),则产率Y为:
(1)
芯材含量测定:精确称量干燥微胶囊质量m3(5g),将其充分研磨后用丙酮超声1h,使囊芯充分溶出,将过滤得到的壁材干燥后称重,得到其质量m4,则芯材含量C为:
(2)
2 结果与讨论
通过单因素试验分析氯化铵用量对自修复微胶囊微观形貌、粒径分布、产率和芯材含量的影响,并在此基础上分析自修复微胶囊的化学结构和热稳定性.表1为1#~5#样品的氯化铵、脲醛树脂预聚体用量及两者的比值.
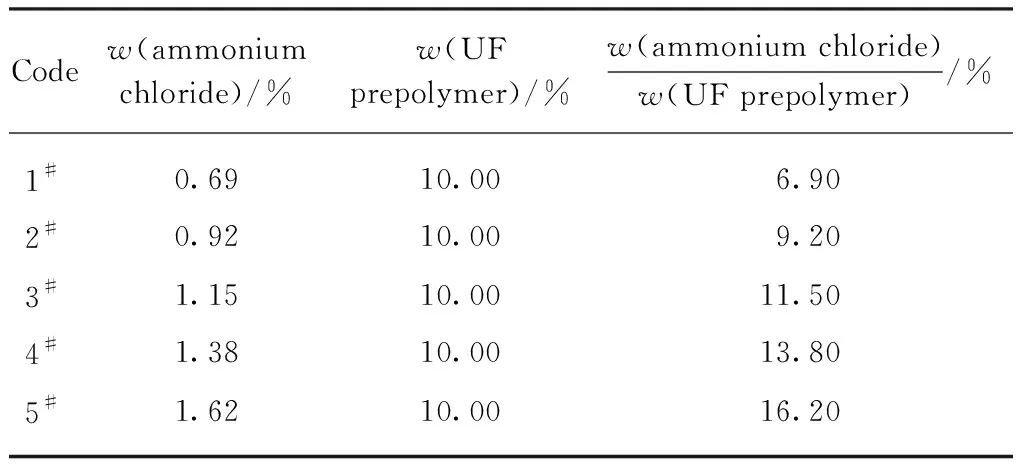
表1 氯化铵及脲醛树脂预聚体用量
2.1 微观形貌
1#~5#样品的ESEM照片见图1~5.由图1~5可见:最终的产物主要是微胶囊和未利用到的壁材颗粒,另外还有少量未被包裹的芯材小液滴附着在微胶囊表面和壁材颗粒上.相较于其他样品,氯化铵用量与脲醛树脂预聚体用量之比为11.50%的3#样品制备效果最好,微胶囊球形饱满,几乎无变形、粘连和破裂,且未利用到的壁材颗粒很少,表面粗糙,整体分散性较好.氯化铵用量与脲醛树脂预聚体用量之比为16.20%的5#样品制备效果差,微胶囊变形、粘连和破裂很多,且未利用到的壁材颗粒很多,表面光滑.与4#,5#样品相比,氯化铵用量与脲醛树脂预聚体用量之比为6.90%的1#样品虽然制备效果较好,微胶囊呈球形,变形、粘连和破裂较少,且未利用到的壁材颗粒也较少,表面较粗糙,但效果依然不如3#样品好.氯化铵用量与脲醛树脂预聚体用量之比为9.20%的2#样品制备效果也较好,但未利用到的壁材颗粒较多,表面较粗糙.
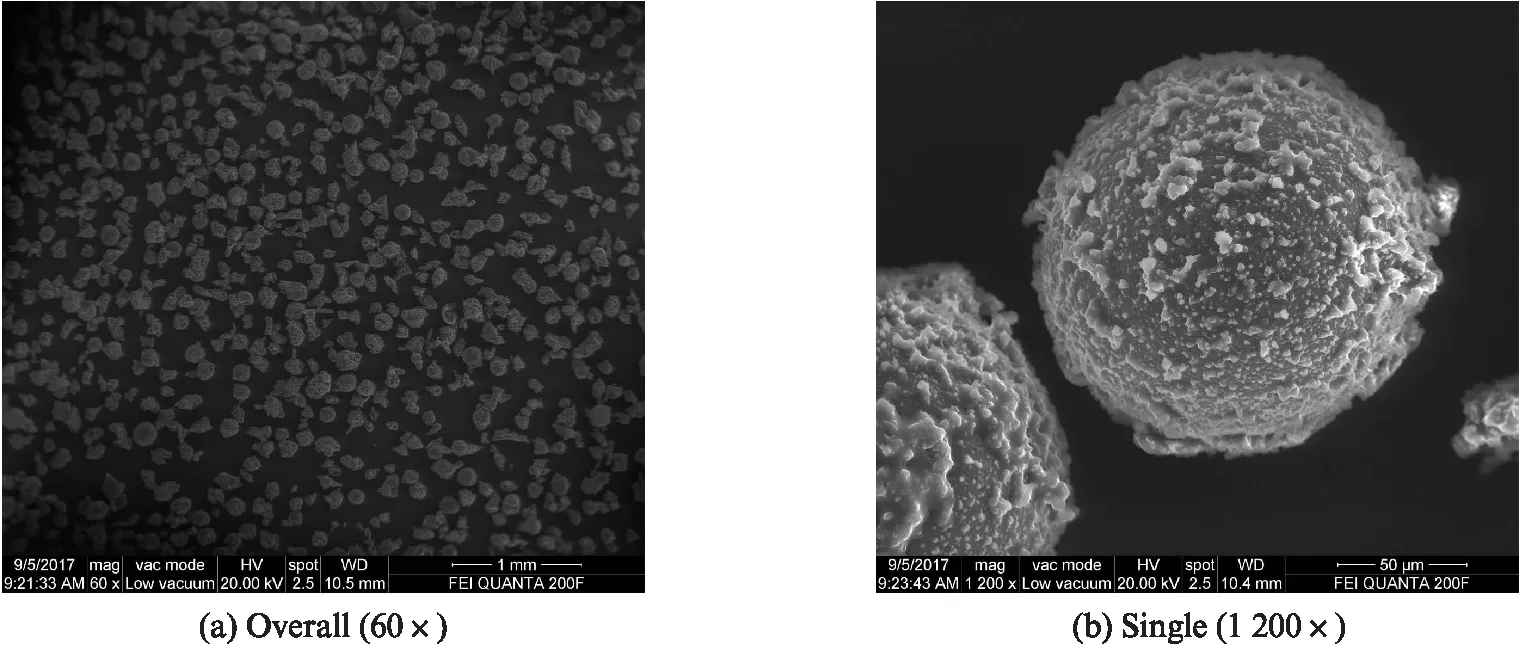
图1 1#样品(氯化铵用量与脲醛树脂预聚体用量之比为6.90%)的ESEM照片Fig.1 ESEM image of sample 1#(w(ammonium chloride)/w(UF prepolymer)=6.90%)

图2 2#样品(氯化铵用量与脲醛树脂预聚体用量之比为9.20%)的ESEM照片Fig.2 ESEM image of sample 2#(w(ammonium chloride)/w(UF prepolymer)=9.20%)

图3 3#样品(氯化铵用量与脲醛树脂预聚体用量之比为11.50%)的ESEM照片Fig.3 ESEM image of sample 3#(w(ammonium chloride)/w(UF prepolymer)=11.50%)

图4 4#样品(氯化铵用量与脲醛树脂预聚体用量之比为13.80%)的ESEM照片Fig.4 ESEM image of sample 4#(w(ammonium chloride)/w(UF prepolymer)=13.80%)

图5 5#样品(氯化铵用量与脲醛树脂预聚体用量之比为16.20%)的ESEM照片Fig.5 ESEM image of sample 5#(w(ammonium chloride)/w(UF prepolymer)=16.20%)
2.2 粒径分布
表2为自修复微胶囊的粒径分布.由表2可见:氯化铵用量与脲醛树脂预聚体用量之比为9.20%的2#样品粒径主要集中于75~150μm;氯化铵用量与脲醛树脂预聚体用量之比为11.50%的3#样品粒径主要集中于75~300μm;氯化铵用量与脲醛树脂预聚体用量之比为13.80%的4#样品粒径分布较为均匀;氯化铵用量与脲醛树脂预聚体用量之比为16.20%的5#样品粒径主要集中于150~600μm;氯化铵用量与脲醛树脂预聚体用量之比为6.90%的1#样品由于在粒径150~600μm中的大部分是未利用到的壁材颗粒,所以其粒径主要集中于75~150μm.

表2 自修复微胶囊粒径分布
2.3 囊化指标
由于部分未利用到的壁材颗粒与微胶囊尺寸大小相近,分筛并不能充分去除多余的壁材颗粒,而未被包裹的芯材小液滴质量很小可忽略不计,所以图6所示的自修复微胶囊产率是混合产物的产率.微胶囊的囊化指标即由混合产物的产率和微胶囊芯材含量共同表征.从图6可以看出:当氯化铵用量与脲醛树脂预聚体用量之比为6.90%~11.50%时,混合产物的产率逐步提升,微胶囊芯材含量也略微增加,所以可以得出此时实际微胶囊的产率是逐渐增加的;当氯化铵用量与脲醛树脂预聚体用量之比为11.50%~16.20%时,混合产物的产率和芯材含量均逐步降低,所以可以得出此时实际微胶囊的产率是逐渐降低的.即氯化铵用量与脲醛树脂预聚体用量之比为11.50%时,混合产物的产率和微胶囊芯材含量均达到最高值,分别为79.05%和68.40%.
2.4 化学结构
将微胶囊充分研磨,再用丙酮超声处理,充分抽提其中芯材,最后烘干得到微胶囊囊壁;按上述微胶囊制备方法,但不加入芯材、跳过乳化阶段,可以获得微胶囊壁材.图7是3#样品、微胶囊壁材、微胶囊囊壁和环氧树脂芯材的红外光谱图.

图6 自修复微胶囊的产率和芯材含量Fig.6 Yield and core content of self-healing microcapsule

图7 3#样品、微胶囊壁材、微胶囊囊壁和环氧树脂芯材的红外光谱图Fig.7 FTIR spectra of sample 3#,microcapsule shell material,microcapsule shell and epoxy resin
由图7可见:微胶囊壁材和微胶囊囊壁的红外光谱曲线形状几乎一致,表明环氧树脂芯材不会参与微胶囊囊壁形成的交联反应而构成微胶囊囊壁的一部分;与3#样品的红外光谱曲线相比,微胶囊壁材和微胶囊囊壁的红外光谱曲线同样存在波数为 1378.67cm-1的C—N伸缩振动峰且在环氧树脂芯材的红外光谱曲线中不存在,环氧树脂芯材的红外光谱曲线存在波数为912.98cm-1的环氧环特征振动峰且在微胶囊壁材和微胶囊囊壁的红外光谱曲线中不存在;这表明脲醛树脂壁材成功包封了环氧树脂芯材,即微胶囊制备成功.环氧树脂芯材的红外光谱曲线中波数为3510.09cm-1的特征峰属于O—H的伸缩振动,而微胶囊壁材和微胶囊囊壁的红外光谱曲线中波数为 3316.90cm-1的特征峰属于N—H的伸缩振动,所以3#样品的红外光谱曲线中波数为 3408.43cm-1的特征峰属于O—H和N—H耦合作用下的伸缩振动,即环氧树脂芯材中O—H和脲醛树脂壁材中N—H耦合,这也证明了微胶囊制备成功.
2.5 热稳定性
图8为3#样品、微胶囊壁材和环氧树脂芯材的TGA曲线;图9,10分别是3#样品在180℃下热处理1,2h后的ESEM照片;图11是3#样品在室温下密封保存8个月后的ESEM照片.
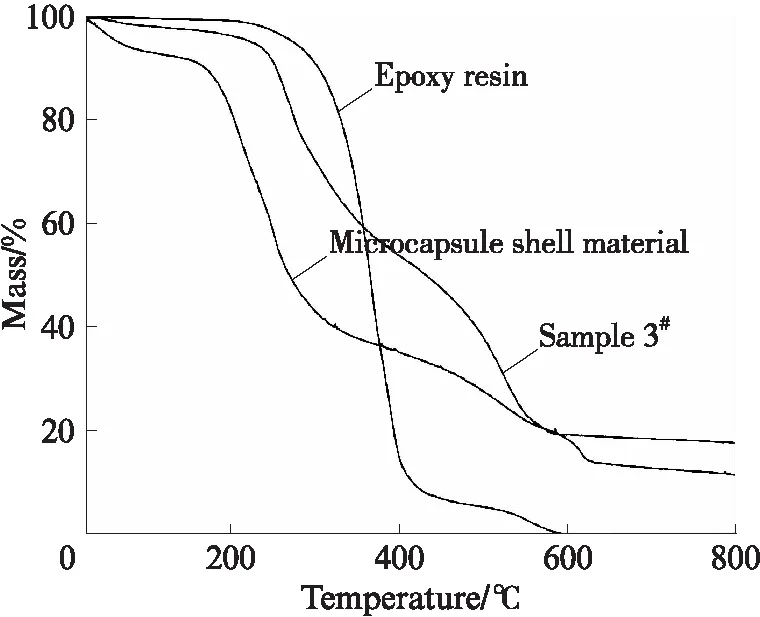
图8 3#样品、微胶囊壁材和环氧树脂芯材的TGA曲线Fig.8 TGA curves of sample 3#,microcapsule shell material and epoxy resin
由图8可见:在0~110℃范围内,微胶囊壁材的质量损失在7%左右,而3#样品和环氧树脂芯材几乎没有质量损失.在0~110℃范围内,由于3#样品和环氧树脂芯材的质量损失很小,此时微胶囊壁材的质量损失是由于样品中残存水分的蒸发引起的;在150℃时微胶囊壁材的质量损失开始逐渐增大,而在230℃时环氧树脂芯材的质量损失开始逐渐增大,表明环氧树脂芯材的热稳定性优于微胶囊壁材;在230℃之后,由于微胶囊壁材的持续分解和环氧树脂芯材的不断自聚,从而导致3#样品的质量直线下降;直至600℃时3#样品的质量损失达80%左右,在此之后3#样品和微胶囊壁材的质量变化不大.
由图9,10可以看出:在180℃热处理温度下,3#样品的整体形貌出现变形和破裂的数量随着处理时间的增加而增多;从微胶囊单个情况可以看出,微胶囊壁材发生分解且微胶囊变形较大,但仍有一部分微胶囊球形良好且饱满.由图11可以看出,自修复微胶囊能在室温中长期稳定存在.
通过自修复微胶囊TGA曲线并结合耐热性分析可知,当温度高于150℃后,微胶囊壁材的质量损失明显,而微胶囊的质量损失并不明显,其原因可能是由于微胶囊中壁材发生分解而环氧树脂芯材没有发生分解,或者是因为环氧树脂芯材的热稳定性优于微胶囊壁材,使得微胶囊的热稳定性得到了提高,并且介于环氧树脂芯材和微胶囊壁材的热稳定性之间.

图9 180℃下热处理1h后的自修复微胶囊ESEM照片Fig.9 ESEM images of self-healing microcapsule at 180℃ for 1h

图11 室温下密封保存8个月后的自修复微胶囊ESEM照片Fig.11 ESEM images of self-healing microcapsule was preserved for 8 months at room temperature
3 结论
(1)当氯化铵用量与脲醛树脂预聚体用量之比为6.90%~11.50%时,混合产物的产率和芯材含量均呈上升趋势;当氯化铵用量与脲醛树脂预聚体用量之比为11.50%~16.20%时,混合产物的产率和芯材含量均呈下降趋势.当氯化铵用量与脲醛树脂预聚体用量之比为11.50%时,混合产物的产率和微胶囊芯材含量均达到最高值,分别为79.05%和68.40%,此时UF/E自修复微胶囊球形饱满,表面粗糙,整体分散性较好,有利于其与水泥基材料的结合.
(2)随氯化铵用量增加,反应体系pH值下降速度加快,加速了脲醛树脂颗粒在微胶囊表面的堆积,使微胶囊的粒径呈上升趋势.在氯化铵用量与脲醛树脂预聚体用量之比为11.50%时,UF/E自修复微胶囊粒径集中分布在75~300μm范围.
(3)通过分析UF/E自修复微胶囊的化学结构和热稳定性,证明脲醛树脂壁材成功包封了环氧树脂芯材,且具有良好的热稳定性,在室温中能长期稳定存在,便于储存.
猜你喜欢
天津科技(2022年7期)2022-07-29 08:42:48
天津科技(2021年7期)2021-07-29 13:47:06
天津化工(2021年1期)2021-01-05 16:42:05
天津科技(2020年7期)2020-07-31 09:10:56
食品安全导刊·下旬刊(2020年3期)2020-07-09 18:55:48
合成树脂及塑料(2020年6期)2020-01-14 01:01:15
中国测试(2018年3期)2018-05-14 15:33:29
江西建材(2018年4期)2018-04-10 12:36:48
武汉工程大学学报(2016年3期)2016-09-27 06:06:07
水生生物学报(2015年1期)2015-02-28 16:00:08
