
基于高通量测序的两种高寒草甸土壤原核生物群落特征研究
2018-12-20 06:57盛玉钰王秀磊李迪强张于光
生态学报 2018年22期
卢 慧,赵 珩,盛玉钰,丛 微,王秀磊,李迪强,张于光,*
1 中央民族大学生命与环境科学学院,北京 100081 2 中国林业科学研究院森林生态环境与保护研究所,国家林业局森林生态环境重点实验室,北京 100091
土壤微生物参与了生态系统的各种重要的生态功能,是评价生态系统各种地下生态与生物地球化学循环过程对环境变化的响应的一个重要指标[1],近年来,已有研究报导了不同环境下土壤微生物的群落组成,有学者认为微生物群落的控制因子会随着研究尺度的不同而发生改变[2]。Zhang等[3]发现土壤微生物群落的分布受到纬度梯度的影响,而更多的研究认为微生物多样性沿海拔梯度的变化不同[4- 5],pH[6]、土壤养分[7]、土壤温度[8]或其他因子也可能是影响微生物群落组成的主要因子。
气候变化对各种生物都会产生很大影响[9]。由于高海拔和极端的气候条件,青藏高原是生态脆弱地区,极易受到环境干扰[4]。高寒沼泽化草甸是青藏高原高寒草甸草地的重要组成部分, 是一种草甸与沼泽之间的过渡植被类型,面积为4.9×104km2,是青藏高原分布面积较广的草地生态系统之一[10]。受到气候暖干化的影响,多年冻土出现退化,使得土壤表层含水量下降,植物群落随之发生变化,从而由高寒沼泽化草甸演替为高寒草甸[11]。有学者认为,植物演替会对土壤微生物的群落结构产生显著影响,甚至还会在很大程度上影响到土壤碳、氮含量的动态变化[12]。因此,作为青藏高原独有的高寒草甸生态系统,研究气候变化对土壤原核生物的影响及其响应是十分必要的,对研究该区域独特的微生物地理区系和预测全球环境变化的影响有重要意义。
近年来,关于高寒生态系统的研究多集中于植被覆盖度[13]、植物群落结构[14]、植物第一性生产力[15-16]、土壤碳循环或氮循环[17-18]等方面,而对草甸生态系统土壤微生物的研究还处于起步阶段。随着分子生物学的发展,高通量测序技术作为研究微生物群落组成的有效手段,已被广泛应用于微生物学的研究领域中,它可以通过对从土壤中提取的微生物总DNA进行测序和比较分析,快速的对微生物进行有效鉴定[19]。目前,由于受到研究技术和分析方法的局限,大部分的微生物多样性研究都集中在物种的多样性和丰富度等方面,没有考虑微生物物种或种群间的相互作用,而分子生态网络(Molecular Ecological Networks)分析方法[20]的构建为预测微生物群落组成和多样性对各种环境变化的可能响应提供了新的机遇。本研究以青藏高原三江源地区高寒沼泽化草甸和高寒草甸的土壤样品为研究对象,通过Illumina Miseq高通量测序和分子生态网络相结合,分析不同高寒草甸类型土壤原核生物群落组成和分子生态网络特征,并探讨影响原核生物群落组成的主要环境影响因子。
1 研究方法
1.1 取样方法
选择青海省三江源地区的高寒沼泽化草甸(Alpine swamp meadow,简称ASM,34°22′15″N, 97°56′57″E,海拔4480 m)和高寒草甸(Alpine meadow,简称AM,35°24′28″N, 99°21′6″E,海拔4140 m)为研究对象,各设立一块样地。每块样地的坡度、坡向、人为干扰情况尽可能一致。为了在考虑空间异质性的同时降低实际地形的取样难度,我们采样时对巢式取样的方法进行了简化,进行了“L”形取样[21]。即在每块样地设立1个200 m×200 m的网格,以网格内任意一个角为起点,在水平和垂直方向上距离分别为10,20,50,100 m和200 m处设置1 m×1 m的样方,每块样地内设立10个1 m×1 m样方,共20个样方。在每个1 m×1 m的样方内采用对角线取样法采集土壤样品,取样深度为0—10 cm,混合后过筛,分成2份,1份低温保存用于土壤理化性质分析,1份-80℃保存用于DNA提取。同时记录采样地点经纬度、地形等。
1.2 土壤理化性质和植物多样性测定
在每个样方中,调查植物种类、多度、高度、盖度等指标,计算重要值。物种多样性的测定采用物种丰富度指数和香农多样性指数来表征。
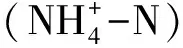
1.3 高通量测序
土壤DNA的提取主要参考Zhou等[23]的方法进行。利用土壤基因组DNA提取试剂盒(MP Biomedical, Carlsbad,CA)先粗提土壤样品DNA,然后利用0.5%的低熔点的琼脂糖凝胶对样品DNA进行纯化,利用NanoDrop ND- 1000分光光度计(Nanodrop Inc)检测样品DNA纯度,若A260nm/280nm>1.80,A260nm/230nm>1.70则表明样品DNA符合要求。最后,利用FlUOstar Optima(BMG Labtechm Jena,Germany)方法对纯化后的DNA进行定量。
以样品DNA为模板,根据16S rRNA的V4高变区设计引物,其正向引物为5′-GTGCCAGCMGCCGCGG TAA- 3′ (515F),反向引物为5′-GGACTACHVGGGTWTCTAAT- 3′ (806R)[24],在PCR反应体系中进行扩增。PCR反应体系参照Ding等[8]的方法。将600 μL的混合液放入Illumina Miseq(Illumina,San Diego,CA)平台(2×150个序列读长)进行测序。
原始序列根据Barcode区分不同的样本序列,利用FLASH算法[25]处理原始序列的前末端和后末端,将测得的序列进行比对分析,去掉低质量、较短序列和无法比对到16S rRNA数据库的序列以保证数据的可靠性。根据UCLUST方法[26],将样点序列以≥97%的相似性划分为1个分类操作单元(Operational Taxonomic Unit, OTU),采用RDP classifier将代表性OTU的序列信息比对至相对应的物种信息[27],最后对所获得的OTU数据进行处理分析。所有的数据分析在美国俄克拉荷马大学环境基因组研究所的网站平台完成(http://zhoulab5.rccc.ou.edu:8080)。
1.4 分子生态网络构建
分子生态网络是基于随机矩阵理论[20],通过在线平台MENA进行构建和数据分析(http://ieg2.ou.edu/mena/),用于研究微生物网络互作关系。为确保OTU相互关系的准确性,在构建网络时,去除同一样地10个平行样品中出现次数低于70%的OTUs。网络构建时,将基于随机矩阵理论,自动生成阈值。每个网络基于快速模块优化的方法形成不同的模块。本研究中,不同网络的拓扑性质通过不同的指数如平均联系数、平均聚集系数、平均路径距离、连通性和模块性[20]来进行表征。网络节点功能通过模块内连接度(Zi)和模块间连接度(Pi)这2个指标来进行划分,将节点划分为4大类型:模块枢纽(Zi>2.5且Pi≤0.62)、网络枢纽(Zi> 2.5且Pi>0.62)、外围节点(Zi≤2.5且Pi≤0.62)和连接节点(Zi≤2.5且Pi>0.62)[8]。此外,将环境因子当成网络节点,与原核生物OTUs一起构建网络,用来研究原核生物网络中的关键影响因子。
1.5 统计分析
采用Excel和SPSS 18.0软件利用对数据进行初步统计和差异性检验;微生物多样性通过香农指数和辛普森指数来表征,并计算微生物的物种丰富度;用除趋势对应分析(Detrended correspondence analysis,DCA)对土壤微生物群落特征进行排序;用典范对应分析(Canonical correspondence analysis,CCA)研究群落分布格局与环境因子的关系;DCA和CCA均使用生物统计学软件R软件中的Vegan软件包[8]进行统计分析。不相似性检验均利用在线网站(http://ieg.ou.edu/microarray/)进行分析。采用SigmaPlot 12.5软件作图。分子生态网络采用Cytoscape 3.0软件进行可视化。
2 结果
2.1 两种高寒草甸的环境因子特征
对样地进行植物调查和植物指标计算,结果表明,ASM(高寒沼泽化草甸)样地的优势种为藏嵩草(Kobresiatibetica)和矮嵩草(Kobresiahumilis);AM(高寒草甸)样地的优势种是矮嵩草和高山嵩草(Kobresiapygmaea)。AM样地的植物多样性高于ASM,而植物丰富度低于ASM(表1)。

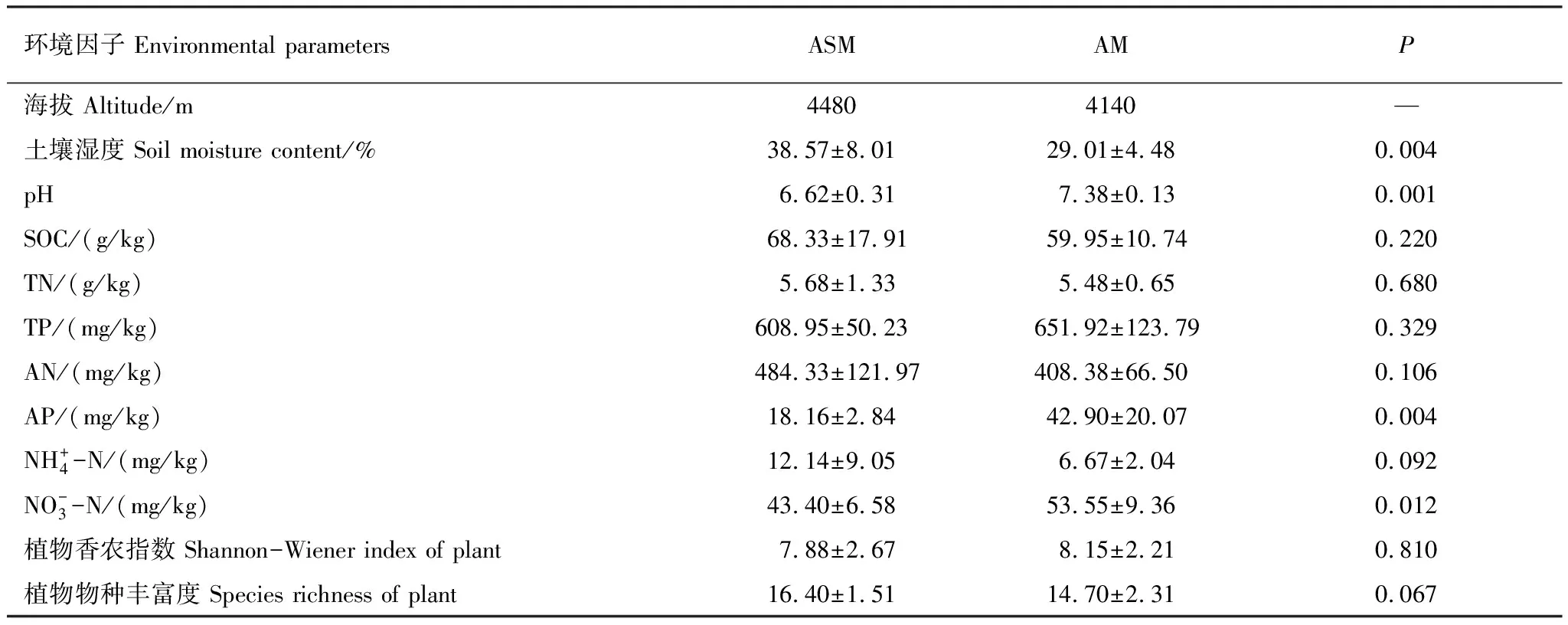
表1 两种草甸类型土壤的环境因子比较

2.2 土壤原核生物的多样性比较
利用Illumina Miseq平台对土壤样品进行高通量测序,按照15000序列重新抽样,基于97%的相似性定位物种水平,共获得23145个OTUs,其中,ASM样地检测到15555个OTUs,AM样地共检测到16225个OTUs。香农和辛普森多样性指数均显示,AM样地的原核生物多样性显著高于ASM样地 (P< 0.1)(表2)。
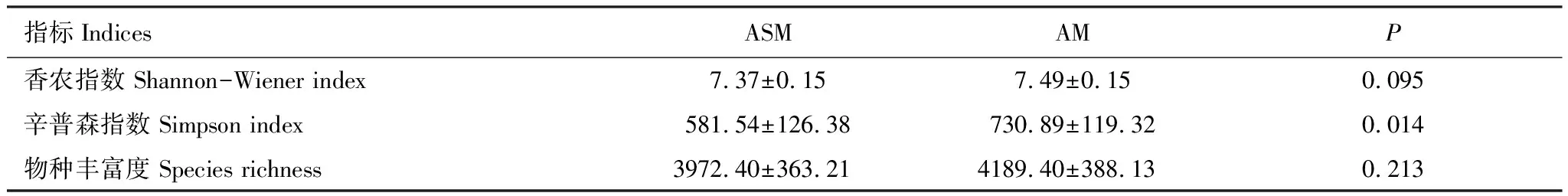
表2 两种草甸类型的原核生物多样性
通过除趋势对应分析(DCA)对原核生物群落组成进行排序,从图1中可以看出,两种草甸类型的原核生物群落能基本分开。进一步进行不相似性检验(包括MRPP、Adonis和Anosim),结果显示两种草甸类型原核生物群落组成差异显著 (P< 0.001,表3)。

图1 土壤原核生物群落组成的除趋势对应分析(DCA) Fig.1 Detrended correspondence analysis of soil prokaryotic community compositionASM:高寒沼泽化草甸,Alpine swamp meadow;AM:高寒草甸,Alpine meadow
Table3Dissimilaritytestofprokaryoticcommunitycompositionbetweentwomeadowtypes

不相似性检验Dissimilarity testδ/R/R2PMRPPδ=0.609 < 0.001AnosimR=0.588 < 0.001AdonisR2=0.183 < 0.001

图2 原核生物群落组成主要门类的相对丰度 Fig.2 Relative abundance in the major phyla of prokaryotic community composition变形菌门,Proteobacteria;酸杆菌门,Acidobacteria;放线菌门,Actinobacteria;拟杆菌门,Bacteroidetes;疣微菌门,Verrucomicrobia; 浮霉菌门,Planctomycetes;厚壁菌门,Firmicutes;绿弯菌门,Chloroflexi;其他,Others;泉古菌门,Crenarchaeota;芽单胞菌门,Gemmatimonadetes
2.3 土壤原核生物的群落组成差异
检测到的OTUs可划分为33个细菌门类和2个古菌门类。其中,变形菌门(Proteobacteria)、酸杆菌门(Acidobacteria)、放线菌门(Actinobacteria)和拟杆菌门(Bacteroidetes)是优势门类(图2),它们在ASM样地中的相对丰度分别为34.8%、24.7%、11.6%和8.7%,在AM样地中的相对丰度分别为32.1%、23.1%、19.5% 和6.5%(图2)。其中,变形菌门、酸杆菌门和拟杆菌门的在ASM样地中相对丰度较高,而放线菌门在AM样地中相对丰度较高。
2.4 原核生物的分子生态网络结构分析
基于RMT算法,利用高通量测序数据构建分子生态网络,比较两种草甸类型土壤原核生物网络联系上的差异,研究它们之间的网络互作关系,网络的拓扑性质见表4。构建的两种草甸类型的网络具有相同的阈值(0.890),且均体现了网络的无尺度特征、小世界特征以及模块化特征。ASM的网络包含739个节点和986个连接数,而AM网络包含860个节点和884个连接数(表4)。比较两个网络的拓扑性质,ASM具有较高的连通性和平均聚集系数,而AM具有较长的平均距离和较高的模块性。
模块枢纽和连接节点是网络联系中的两大重要节点。从图3可以看出,ASM含有6个模块枢纽,其中,4个模块枢纽属于变形菌门,1个属于绿弯菌门以及1个属于浮霉菌门。而AM含有5个模块枢纽,2个属于放线菌门,1个为变形菌门, 1个为疣微菌门以及1个为芽单胞菌门。ASM含有5个连接节点,而AM中不含连接节点。
2.5 影响土壤原核生物群落组成的环境因素

通过构建网络的方法进一步进行分析,比较原核生物群落组成与环境因子的联系紧密程度,结果显示,土壤pH值在所检测的环境因子中具有最高的连接数目,再次表明了pH值在高寒草甸原核生物群落组成上产生了极其重要的作用(图5)。

表4 两种草甸类型的原核生物网络关系的拓扑性质

图3 基于网络拓扑性质的原核生物网络Z-P分布图 Fig.3 Z-P plot showing node categories distribution of prokaryotic network based on their topological properties

图4 原核生物群落与环境因子间的典范对应分析 (CCA) Fig.4 Canonical correspondence analysis of prokaryotic community with environmental factors

图5 原核生物物种组成与环境因子的网络互作关系Fig.5 Network interactions between environmental factors and prokaryotic taxonomic community OTUs绿色的点表示与pH直接连接的OTUs,蓝色的点表示与pH间接连接的OTUs
3 讨论与结论
Illumina MiSeq高通量测序技术的发展提供了一个强大的、高效的平台,可以帮助我们迅速和有效地确定微生物的群落结构组成[18]。近年来,越来越多的学者关注于土壤微生物优势菌群的研究,有学者认为,微生物群落的整体组成在不同生境中的差异可能较大,但优势菌群基本相似,如Shen等[28]通过高通量测序技术对长白山地区土壤微生物的研究发现,变形菌门、酸杆菌门和疣微菌门是微生物主要类群,相对丰度分别为23.1%、20.8%和17.29%。Zhang等[7]对高寒草甸演替过程中微生物群落变化进行了研究,结果表明酸杆菌门、变形菌门、放线菌门和浮霉菌门是优势菌群。本研究结果表明,两种草甸类型的原核生物多样性有显著差异,而变形菌门、酸杆菌门、放线菌门和拟杆菌门的是两种高寒草甸类型中相对丰度最高的重要细菌类群,在两个样地中相对丰度累计均超过79%。可以看出,土壤微生物优势菌群在不同土地类型和不同海拔梯度都具有一定的相似性,这可能是由于这些微生物类群种类最多、分布广,且个体小、扩散性强,易形成随机性和广泛性的分布特点[29],除此之外,微生物生境特异性低,适应环境能力强可能也是一个重要因素[30]。
分子生态网络作为一种功能强大的工具,可以提供不同物种之间复杂的生态相互作用的重要信息,并揭示微生物结构网络拓扑结构的变化[31]。ASM网络较短的平均路径距离和较高的连通性表明其土壤原核生物之间的相互作用比AM网络更复杂、更紧密。然而,较短的平均路径距离意味着局部的干扰可能会更快地传达到整个网络[2,32],而紧密连接的群落可能对干扰更加敏感[2],这意味着具有较长路径距离的AM网络较ASM网络更加稳定。ASM网络的模块枢纽种类较为单一,大部分OTUs属于变形菌门,而群落组成的单一化可能导致其联系较为紧密,对外界的抵抗能力反而较弱[33]。此外,模块性的比较结果也证实了这一点。网络模块性作为网络系统对抗外界变化的抗性指征[8],在AM样地中较高,在ASM样地中较低,说明AM网络在应对外界环境变化时将具有更高的抗性。
土壤pH值通常是影响微生物多样性的重要因子之一[6],它与土壤微生物群落结构和组成有着极为紧密的关系,这种相互关系在在不同空间尺度的研究中都有报道,如Shen等[6]发现土壤 pH 值是控制土壤微生物多样性和群落组成的关键性因子,并指出pH值对微生物的分布普遍存在影响;张于光等[34]认为pH值不仅是影响土壤微生物群落结构的主要因素,而且影响了微生物的海拔分布格局。土壤pH值是一个复杂的环境参数,有学者认为,土壤pH值之所以能对微生物的群落结构产生重要影响,可能与土壤pH值能够对土壤其他因子的变化产生间接的影响有关[35]。本研究中,CCA和分子生态网络的分析结果均表明,pH值是影响高寒草甸土壤原核生物群落特征的主要因子。
青藏高原草地生态系统在全球气候变化中扮演着重要角色,对全球气候变化十分敏感。本文通过对青藏高原特有的代表性的植被类型高寒沼泽化草甸和高寒草甸进行研究,分析其地下原核生物在全球气候变化下的响应,以期为高寒草甸生态系统的保护性管理和应对气候变化提供一定的理论依据。
猜你喜欢
成都信息工程大学学报(2022年4期)2022-11-18
青海草业(2022年2期)2022-07-23
中国比较医学杂志(2020年4期)2020-05-26
水生生物学报(2019年4期)2019-07-20
活力(2019年21期)2019-04-01
生物安全学报(2019年3期)2019-02-15
川北医学院学报(2019年6期)2019-02-10
河北林业科技(2016年5期)2016-11-08
安徽医科大学学报(2015年9期)2015-12-16
动物医学进展(2015年10期)2015-12-07
