
燃料电池低Pt核壳结构催化剂的最新研究进展
2018-12-13 07:38余锦南廖世军
中国材料进展 2018年11期
余锦南,廖世军
(华南理工大学化学与化工学院 广东省燃料电池技术重点实验室,广东 广州 510641)
1 前 言
自从工业革命以来,人类主要采用燃烧的方式利用自然能源,随着人类对能源需求的日益增长,能源资源问题以及由于大量燃烧矿物燃料所引起的环境污染问题变得日趋严峻,迫使人类社会努力寻找更为高效、清洁的能源利用方式。燃料电池作为一种可直接将燃料的化学能转化成电能的电化学装置,具有能量转换效率高、零排放或者低排放等重要优点[1, 2],被誉为21世纪最为重要的新能源技术。发展燃料电池技术,是解决人类目前面临的环境与能源问题的重要途径之一。
低温质子交换膜燃料电池是最有可能在短期内实现大规模应用的一种燃料电池技术,也是最有可能应用于电动汽车的一类燃料电池,世界上各发达国家在低温质子交换膜燃料电池技术方面已开展了大量的研究、开发和示范工作。然而,其大规模商业化仍然步履蹒跚。影响燃料电池大规模商业化的主要原因包括:使用贵金属Pt作为催化剂而导致的高成本,以及催化剂耐久性不足的问题。
燃料电池阴极氧还原反应缓慢的动力学过程是限制燃料电池效率的主要原因之一[3-5]。阴极氧还原反应催化剂以Pt基催化剂最优,而贵金属Pt储量少、价格昂贵,这是导致燃料电池高额生产成本的主要原因[6, 7]。此外,Pt易受到一氧化碳、甲醇、甲酸等物质的毒化,类似高压操作等条件也易引起燃料电池寿命的衰减。因此,开发具有高活性和低成本的纳米催化材料对燃料电池的发展有非常重要的意义。
当前,尽管报道了许多廉价的非Pt燃料电池催化剂[8-11],但其活性和酸性环境下的稳定性尚达不到实际应用的要求,Pt及其合金依然是最有效的燃料电池阴极氧还原反应催化剂。Pt与其他贵金属Pd[12, 13]、Ir[14]、Ru[15]、Rh[16]、Au[17]等或者过渡金属Ni[18]、Co[19]、Fe[20]形成的合金作为催化剂已经引起了广泛的关注。然而,PtM(其中M为Pt以外的金属)合金催化剂中Pt的利用率不高、金属M在酸性介质中稳定性较低以及PtM合金在燃料电池工作电势下不稳定等,这些因素都限制了它的商业化生产应用。
当Pt分散于传统载体上时,仅有表层的Pt原子暴露在电解质中,因此仅有部分的Pt被利用。例如,3 nm的Pt纳米粒子,只有30%的Pt得到有效利用,其余70%的Pt则分布在颗粒内部而无法参与催化反应[21]。而Pt基核壳催化剂,是以其他贵金属或过渡金属及其化合物为核,通过化学键或其他作用力,在核表面形成数个Pt原子层的纳米催化材料。由于Pt仅分布在催化剂粒子的壳层,因此Pt基核壳催化剂可大大降低Pt的用量,提高Pt的利用率。研究表明,低Pb核壳催化剂的质量活性可高达商业Pt/C的数10倍[22]。
2 基于贵金属为核的核壳结构催化剂
基于晶格错配的现象,当Pt沉积在另一个基底上时会产生不同的拉伸或伸缩应力,同时还可以调控其电子结构,从而提高催化剂的活性和稳定性[23]。以其它价格相对低廉的贵金属作为核开发低Pt核壳结构催化剂,在降低金属Pt用量的同时,可实现催化剂更高的活性和稳定性,缓解当前因Pt储量少、价格昂贵等问题导致的燃料电池发展缓慢的困境。目前研究比较多的贵金属核有Pd、Au、Ru、Ir、Ag及其合金,相较于贱金属,它们具有更好的热力学稳定性,同时也不易溶出。通过控制核纳米粒子的形貌、形成方式及Pt壳层的厚度等,均可改善核壳催化剂的性能。
2.1 以Pd为核的核壳结构催化剂
Yu课题组[24]利用Br-作为模板剂引发PtCl62-与Pd纳米线的置换,从而形成壳层薄、表面光滑、超细均匀的Pd@Pt核壳结构纳米线。由于Br-与PtCl62-形成的中间体PtBr62-具有较低的氧还原电位,从而降低了反应速率,以便Pt可以完整地包覆于Pd纳米线,形成光滑的Pt壳层。完整的Pd@Pt纳米线核壳结构有助于提高材料的活性和稳定性。值得注意的是,Pd@Pt的初始质量活性比商业Pt/C高2.45倍,而在经过60 000圈循环后,它的质量活性是同等情况下商业Pt/C的8.5倍。
Xia课题组[25]利用配体效应和应变效应的组合原理使Pd、Pt之间的晶格失配,制备了Pd@Pt核壳结构催化剂。如图1所示,通过制备多重孪晶Pd十面体,再将Pt原子沉积于其表面。如图1b和1c所示,Pt原子优先沉积在十面体的顶点,随后沿着其边缘/脊方向沉积扩散至其他区域,最后沉积至整个面。由于十面体Pd边缘的(100)和(211)晶面更易捕获Pt,最终所得的催化剂为以凹面Pt为壳层的十面体核壳催化剂。与商业Pt/C相比,Pt的含量为29.6%的凹面十面体表现出更为出色的氧还原催化活性和稳定性。在催化剂制备过程中,使用如聚乙烯吡咯烷酮(PVP)等稳定剂能更好地形成核壳结构的催化剂,但最终所得催化剂表面通常也会吸附上稳定剂。而催化剂表面稳定剂的存在,会覆盖了催化剂的活性位,从而限制了催化剂的活性。鉴于此,他们利用了一种无需使用稳定剂的方法,制备出表面含有3~4个Pt原子层的八面体Pd@Pt3~4L核壳结构催化剂[26]。他们将事先制备的八面体Pd分散在碳载体上以达到稳定核的作用;随后通过调节反应温度和前驱体Pt(II)的浓度,抑制Pt颗粒在八面体Pd上的形成,实现Pt在八面体Pd表面的薄层沉积。他们对比了使用PVP作为稳定剂制备的Pd@Pt的性能,发现不使用稳定剂制备的催化剂具有更高的氧还原催化活性,其活性更是高于商业催化剂的4倍之多。此外,凹面体Pd@Pt体系在甲醇氧化方面也具有很好的活性。

图1 十面体Pd及其褶皱边示意图(a),Pt原子沉积在Pd十面体(b)和二十面体(c)过程的示意图 [25]Fig.1 Schematic of a decahedron viewed along its 5-fold axis, showing the type-A and type-B vertices, as well as the ridges and edges(a); schematic illustrations showing how the Pt atoms are deposited onto Pd decahedral (b) and icosahedral (c) seeds, respectively, and the possible routes for the initially deposited Pt atoms to diffuse across the surface[25]
Guo课题组[27]利用一步法制备出均匀的、具有超薄Pt壳层的Pd@Pt凹面体核壳结构催化剂,该催化剂表现出了超高的甲醇氧化活性。该研究表明,合金体系中的Fe3+、金属前驱体中的acac-和抗坏血酸之间的协同调控作用,对制备凹形多面体Pd核尤为关键。通过考察在不同反应时间下纳米粒子的结构和形貌,他们发现制备Pd@Pt凹面体的过程可分成以下两步:1) Pd凹面体的形成;2) Pt沉积在Pd上形成Pd@Pt凹面体核壳结构催化剂。研究表明,该Pd@Pt催化剂在电催化甲醇氧化和催化硝基苯加氢反应中都表现出优于商业Pt,Pd催化剂的超高活性。
通过改变Pd@Pt催化剂表面结构与应力也是提高其氧还原催化活性的方法之一。Wu等[28]开发出了一种简单的晶种法,可使薄层Pt生长在Pd二十面体、八面体和四方体表面。由于Pt的表面能高于Pd,当Pt沉积于Pd时通常更倾向于岛状生长(如Volmer-Weber模式)沉积,以达到热力学的动态稳定,由此导致Pt以颗粒状的形式生长在Pd表面。而这种方法的关键在于利用油氨中的胺基团对Pt表面进行热力学调控,在胺的强吸附作用下可降低Pt的表面能,从而使其在Pd表面以层状沉积。二十面体的Pd@Pt与八面体、立方体的Pd@Pt以及商业Pt/C相比,其氧还原催化活性均有显著提高。通过几何相分析,他们认为二十面体的Pd@Pt催化剂中的大部分Pt壳层同时受到压缩与拉伸应力的作用,由此导致了它们在暴露同等的{111}晶面的条件下,具有更高的氧还原催化活性。
Shao课题组[29]利用以柠檬酸控制Pt壳层生长的策略,制备出克级的Pd@Pt/C核壳结构催化剂。如图2所示,他们通过考察在不同反应时间下所得的Pd@Pt/C中,Pt的沉积量和Pd的损失量的关系,推测形成该类型的核壳催化剂先后需要3个步骤:1)因Pd/PdCl4-(0.591 V)与Pd/PtCl4-(0.755 V)电对的标准氧化还原电势差引起的置换反应;2)柠檬酸将PtCl4-还原成Pt;3)Pt离子通过瞬间吸附在Pd表面而引起电子从Pd向Pt离子转移从而将其还原。研究表明,柠檬酸的存在对在Pd表面形成完整Pt壳层起到关键作用。当该制备条件缺少了柠檬酸时,Pt在Pd表面形成3D核或团簇,而这种类型的Pd@Pt/C催化剂中Pt的质量活性往往低于完整壳层的Pd@Pt/C催化剂。此方法制备的Pd@Pt/C质量活性是商业Pt/C的4倍。

图2 随着合成反应的进行,利用电感耦合等离子体质谱(ICP-MS)所测得的金属Pd与Pt的含量变化[29]Fig.2 Metal loadings (wt%) measured from ICP-MS of Pd@Pt/C catalysts at different reaction time during the spontaneous deposition of Pt on Pd nanoparticles. The dashed and dotted lines represent the theoretical Pd wt% calculated from the measured Pt wt% with and without involving Pd dissolution during Pt deposition[29]
2.2 以Ru为核的核壳结构催化剂
在Pt表面,甲醇氧化的步骤包含:1)甲醇吸附;2)C—H键活化(甲醇分解);3)水吸附;4)水活化;5)CO氧化[30]。对于纯Pt催化剂,甲醇氧化的中间产物CO会覆盖在活性位点上从而使催化剂活性下降,但当向催化剂中引入第二组分金属元素时,这个现象就会被改善。而研究证明Ru对于提高Pt基催化剂甲醇氧化活性是最有效的。它的机理如下:
Pt+CH3OH→PtCOads+4H++4e-
(1)
Ru+H2O→Ru(OH)ads+H++e-
(2)
PtCOads+Ru(OH)ads→CO2+Pt+Ru+H++e-
(3)
由此看出,Ru是很好的抗CO中毒的催化剂。通过改善Pt-Ru催化剂的组成、结构、形貌、粒径大小均可以改善催化效果。鉴于此,Ru@Pt核壳结构催化剂可以用于甲醇氧化[31-33]、甲酸氧化[34]及氧还原反应等[34]。
Chen课题组[31]制备了近乎单原子层Pt包覆的Ru@Pt核壳结构催化剂。相比于纯Pt及PtRu合金催化剂,Ru@Pt催化剂节约了60%的Pt用量,其催化甲醇燃料电池的功率密度增长了450%(从17.1增长到92.8 mW·cm-2·mg-1),同时开路电压也增长了0.18 V。该研究表明,性能如此大幅度的增长得益于Pt表面受到的压缩应力。此应力会引发Pt表面电子向Ru转移,从而提高了催化剂的表面活性吸附和甲醇电氧化活性,并且加速一氧化碳氧化,直接促进了阳极反应。
近期Jaramillo研究组[35]基于一种湿化学合成法,系统地研究了Ru@Pt/C核壳结构催化剂的制备过程。他们通过调节Ru、Pt比例和催化剂的预处理等制备参数,优化出可制备较薄Pt壳层的条件,进而使该Ru@Pt/C型催化剂的氧还原催化活性有了显著的提高。0.9 V时的质量活性为0.50 A/mg Pt,经过30 000圈循环后,Ru@Pt/C显示出良好的电化学稳定性,其质量活性可达初始值的85%。
2.3 以Ir为核的核壳结构催化剂
密度泛函理论(density functional theory, DFT)计算表明,在Ir表面形成单层Pt(PtML/Ir)比PtML/Ru具有更优的结合能[36]。由此推断,Ir@Pt类型的催化剂亦可获得较为可观的氧还原催化活性。鉴于此,作者团队利用脉冲电化学沉积的方法,在Ir纳米粒子表层沉积薄层的Pt壳层,制备出一种具有核壳结构的Ir@Pt/C催化剂[37]。该催化剂在电催化甲醇氧化反应和氧还原反应中表现出优异的性能,其质量活性可达合金PtIr/C和商业Pt/C催化剂的数倍。另外发现,Ir@Pt/C催化剂中Ir 4f的结合能相较于Ir/C中Ir 4f发生了负移,而其中Pt 4f的结合能又相较于Pt/C中Pt 4f的发生了正移。这可能是由于Pt与Ir原子轨道间的相互作用,导致电子从Pt原子轨道向Ir原子轨道转移所引起的。作者认为,Ir和Pt之间电子的相互作用,可能是Ir@Pt/C催化剂相较于Pt/C和PtIr/C具有更低的一氧化碳起始氧化电位的重要原因。而Pt与Ir之间的协同作用和高分散的Pt是使得Ir@Pt/C核壳催化剂具备高活性的重要因素。此外,作者团队也研究了利用脉冲电化学沉积的方法在Ir/C膜电极(memberane electrode assembly, MEA)上原位制备Ir@Pt/C核壳结构催化剂,该方法制备的Ir@Pt/C-MEA在燃料电池系统测试时也表现出优异的性能[38]。
Jaramillo课题组[39]利用化学还原法,先制备出均一的Ir纳米溶胶,然后将Pt还原在Ir的表层,制备出高活性和稳定性的Ir@Pt/C核壳结构催化剂。经循环10 000圈后,其质量活性是商业Pt/C的1.8倍。Ir@Pt/C催化活性的提高与Ir与Pt的相互作用有密切联系。研究表明[40],考察循环伏安曲线中*OH的脱附峰位移情况可得知Pt—O结合能的变化信息。他们发现,Ir@Pt/C的*OH脱附峰相较于Pt/C的正移了30 mV,这表明前者中的Pt-Ir间的相互作用使其比后者具有了更弱的Pt—O键,表明前者具备更高的氧还原催化活性。此外,通过改善Ir@Pt催化剂的粒子分布、壳层沉积的均匀性,以及减少壳层的厚度和几何形状均可以使其催化性能得以提升。
2.4 以Au为核的核壳结构催化剂
除Ru,Ir外,Au也可以提高Pt基催化剂的抗中毒能力,因此Au@Pt型核壳结构催化剂也受到广泛关注。但Au@Pt催化剂普遍的问题是制备过程中极易团聚。针对这个问题,Zeng等[41]利用置换反应成功制备了单分散的Au@Pt核壳结构催化剂。将其负载于碳粉后,该催化剂表现出超高的甲醇氧化催化活性。由于Pt层下Au的存在,使Pt与Au之间存在电子交换,促使了活性氧物种在Pt表面的形成,由此促使Au@Pt/C结构催化剂表现出超高的活性。而Pt表面的活性氧物种又可帮助除去类似CO的中间产物,从而提高了催化剂的活性。Cui等[17]利用欠电位沉积技术制备了Au@Pt核壳结构催化剂,并将其负载在石墨烯上,该催化剂亦表现出很高的氧还原和甲醇氧化催化活性。
Wang课题组[42]用湿化学法在MoS2上合成具有3D结构的Au@Pt纳米枝晶核壳结构催化剂。他们先将Au负载于MoS2上作为晶种,然后通过调节H2PtCl6的浓度、反应时间和温度以控制Pt壳厚度。他们发现Pt壳层越薄,其电化学比表面积越大,这也越有利于甲醇氧化反应。由于MoS2的大比表面积、Au@Pt的独特多孔纳米结构以及MoS2与Au@Pt之间的协同效应,使得该催化剂比传统的Pt/C催化剂表现出了更好的电催化活性和稳定性。
Chen课题组[43]直接利用自发沉积反应在Au上沉积一个原子层厚的Pt层。通过调节温度和Pt前驱体浓度,可以控制Pt在Au表面的均匀分布。所得Au@Pt/C核壳结构催化剂有着优异的氧还原催化活性和稳定性,其质量活性是Pt纳米粒子的3.5倍,且经过长时间循环也没有明显失活。
通过在纳米颗粒表面外延生长Pt层,从而制备出具有孪生结构和规则形貌的低Pt核壳结构催化剂,能有效提高催化剂的氧还原催化活性稳定性。然而,由于Pt具有较高的表面能和较强的内应力,通常很难形成稳定、多重的孪生结构。且由于孪晶界具有很高的能量,这些孪生结构通常会在氧还原过程中被逐渐腐蚀,进而导致活性下降。鉴于此,Zhang课题组[44]开发了一种Pt层外延生长于Au核的五角星型十面体Au@Pt核壳结构催化剂。在合成过程中,他们利用胺基与Pt的强吸附作用以降低Pt的表面能,从而促进了Pt在Au核表面的外延生长。同时合成体系中的Br-也很大程度上稳定了Pt的{111}晶面,有效地防止了在Au表面形成Pt颗粒。该催化剂表现出了超高的氧还原催化活性和循环稳定性,质量活性高达0.94 mA/μg Pt。
2.5 以Ag为核的核壳结构催化剂
面心立方结构(fcc)Ag的晶格间距为0.409 nm,这与fcc结构Pt的值非常接近,因此Pt很容易在Ag上沉积[45]。此外Ramirez-Caballero等[46]通过DFT计算也证明了Ag@Pt型核壳结构有相当高的稳定性。Li等[47]利用非离子型表面活性剂在室温下合成了Ag@Pt核壳结构催化剂,其中Pt是以枝晶状包覆于Ag核表面,该催化剂表现出了优异的抗CO中毒性能。Yu等[48]用两步法制备出约15 nm大小的Ag@Pt核壳结构纳米粒子,将其负载于碳纳米管后,该催化剂表现出了较好的氧还原催化活性。Chen等[45]利用甲醛和乙二醇作为还原剂制备出的Ag@Pt/C核壳结构催化剂,表现出了超高的甲醇氧化催化活性和氧还原催化活性。
核壳结构催化剂的合成通常是采用种子生长法,即以金属核纳米粒子作为种子,用化学还原法将壳层金属前驱体还原沉积于种子上形成薄壳层[49-51]。这种方法有两个明显缺点,首先是材料的可放大性不足。为减少不可控的壳层材料团聚等副反应发生,需将壳材料前驱体的浓度控制在非常低的水平,也即需在强还原剂的存在下控制前驱体的加入速度。这样的方法可成功地制备出小剂量的Ag@Au、Pd@Pt和Ag@AgPd等材料。然而,当按比例放大时,同样的催化剂却无法得到[52-54]。另一个问题是核金属与壳金属之间的置换反应会影响纳米粒子的结构[55]。Linic课题组[56]利用原位化学修饰前驱体的方法,合成了单层Pt原子完整包覆、同时又可大量制备的立方体Ag@Pt核壳结构催化剂。研究表明,通过对PtCl42-前驱体进行特定的配体更换,可降低Pt2+前驱体的还原电位,从而遏制了如Pt的均相成核及Ag与Pt置换等副反应的发生。这表明该合成反应对反应物的浓度相对不敏感,且易于放大。他们在相同体积的反应容器中,将底物放大了10倍,依然可得到10倍的产物,证明了该反应的易于放大的特性,也为M@Pt类催化剂的放大生产提供了新思路。
3 基于金属合金为核的核壳结构催化剂
以金属合金为核沉积Pt所制备的核壳催化剂,其性能较单金属沉积Pt有更大优势。如以PdCu为核的低Pt核壳催化剂,Pd与Pt之间仅有0.77%的晶格失配,而Cu加入Pd核中可改变核的晶格参数,使得核与Pt层因晶格失配产生应力。此外,Cu是具有较正的可逆电位的过渡金属,这使催化剂在酸性介质中相对稳定[57]。
同样地以PdCu2为核,Yoo等[58]所报道的具有中空结构的PdCu2@Pt/C型核壳结构催化剂也表现出较好的氧还原催化活性和稳定性。其中核PdCu2用化学还原的方法制备,并通过热处理使其形成有序的金属间化合物。而Pt壳层则是通过置换反应沉积到PdCu2核上。他们发现,在酸性和低Cl-浓度的制备条件下,可在保持有序的金属间化合物核的同时,形成具有中空结构和均匀Pt壳层的PdCu2@Pt核壳结构催化剂。
研究发现,通过调节Pt表面受到的压缩应力来优化Pt—O结合能可以有效地调节氧还原催化性能[59, 60]。相反,通常Pt(111)面受到拉伸应力会增强Pt—O键,而这对氧还原反应是不利的。然而,在Huang课题组[22]制备的PtPb@Pt核壳结构纳米片催化剂中,由于顶部及边缘的Pt(110)受到较大双轴拉伸应力,导致一些表面的Pt处于轻度压缩状态,从而优化了Pt—O的结合能,使得催化剂拥有超高的氧还原以及甲醇、乙醇氧化活性。在强双轴拉伸应力作用下,其质量活性比商业Pt/C提高了26.9倍,并且在循环了50 000圈后性能依然不衰减,形貌组成依然无明显变化。在此之前,尚未有其他研究人员报道过强拉伸应力对催化剂氧还原催化活性有促进作用。
作者团队[61]采用欠电位沉积技术制备了Pd1Ru1Ni2@Pt/C纳米粒子,其氧还原质量活性是商业Pt/C的3.3倍。还研究了Pd,Ru,Ni的不同原子比对所得催化剂性能的影响,得到最优原子比为1∶1∶2。且Pd1Ru1Ni2@Pt/C相比于Pd1Ru1@Pt/C,其活性会有明显的提高。作者分析认为是因为Ni在反应过程中溶解从而使催化剂形成多孔结构,且残留的Ni对催化剂的性能也有促进作用。
Adzic课题组[62]制备了一种新型Ti-Au@Pt核壳结构催化剂,其边与顶点覆盖TiO2,而Pt层则沉积在核表面无氧化物覆盖区域,边与顶点覆盖氧化物使表面的Pt被分隔。该催化剂氧还原质量活性比商业Pt/C高13倍,且在循环10 000圈后,活性依然不衰减。他们认为,此催化剂的高活性与稳定性得益于TiO2对表面Pt的稳定作用。边缘上的Pt极易溶解,而边及顶点上的TiO2则有效地防止溶解。此外,Au对Pt的修饰也是提高催化剂活性的重要因素。
Yoon等[12]利用简易的一步合成方法,制备了一种Mo掺杂的八面体PdPt@Pt核壳结构催化剂。其壳层由2~3个Pt原子层构成,而Mo则掺杂于该壳层的顶点和棱边上,这使其在表面形成了超高氧还原催化活性位点。
为避免催化剂表面吸附了表面活性剂而导致活性下降,Song等[63]在无表面活性剂存在的情况下,利用液相合成法,以抗坏血酸还原K2PtCl4、K2PdCl4及NaAuCl4,制备出具有超薄Pt层(小于1 nm)的Pd3Au@Pt核壳催化剂。通过XPS分析发现,Pd3Au@Pt中Pt 4f7/2峰发生了负移,而Pd 3d5/2峰和Au 4f7/2峰均发生了正移,这表明Pt与核中的Pd和Au发生了电子转移,部分电子从核转移到了壳层的Pt中。DFT计算表明,壳层Pt因带有更多的负电荷而具有更好的氧还原催化活性[64]。
有研究证明,尽可能地减少Pt表面的粗糙程度有利于提高其在氧还原反应中的活性[65, 66]。近年来研究表明,欠电位沉积可有效地在Pd纳米线上沉积薄层Pt。所得到的Pd@Pt催化剂也表现出很好的氧还原催化活性与稳定性。这很大程度上得益于其一维薄层结构以及光滑的表面[67, 68]。利用这种方法和理论,Sun等[69]制备了以FeNiPt合金为核、Pt为壳层的FeNiPt@Pt核壳结构催化剂。电化学测试结果显示,在同样组成条件下,具有光滑Pt壳层的FeNiPt@Pt催化剂性能要优于壳层为Pt粒子的FeNiPt@Pt催化剂,且该光滑壳层的FeNiPt@Pt催化剂无论相比商业Pt/C还是FeNiPt合金,性能都有很大提升。
4 非金属为核的核壳结构催化剂
以贵金属为核的低Pt核壳结构催化剂,虽然降低了Pt的用量,但材料成本并没有降低到合理的水平。此外,在酸性条件下,过渡金属的溶解,也是导致催化材料性能衰减的原因之一。因此,开发以非金属为核的低Pt核壳结构催化剂,有利于在降低燃料电池催化剂成本的同时,提高催化剂在酸性条件下的催化性能与稳定性。
因具有良好的导电性、电化学稳定性以及耐腐蚀性等特点,过渡金属氮化物(transition metal nitrides,TMNs)近年来已成为核壳催化剂核材料的研究热点。作者团队[70]采用络合-氮化工艺成功制备了颗粒尺寸小于10 nm的多组分氮化物纳米粒子。随后利用脉冲电沉积技术将Pt沉积到氮化物表面,制备出TiMN@Pt型核壳结构催化剂,如图3a所示。该催化剂的质量活性可达商业Pt/C的5倍,在性能方面完全媲美以贵金属为核的催化剂,但却具有更低的成本和更好的稳定性。进一步研究发现,在氮化钛中掺杂微量Ni元素,能够进一步提升催化剂的活性与稳定性。但是仍然有不足的地方是TiMN纳米粒子依然过大,且分散性不好,为了进一步改善氮化物为核的催化剂结构,作者团队利用氮掺杂的碳纳米管(NCNTs)对氮化物进行分散负载[2]。通过这个方法,成功地制备了高分散且粒径较小(5 nm左右)的Ti0.9Cu0.1N纳米粒子。利用脉冲电沉积技术在Ti0.9Cu0.1N/NCNTs上沉积Pt,而Pt能选择性地沉积在氮化物表面形成Ti0.9Cu0.1N@Pt/NCNTs催化剂,如图3b所示。电化学测试结果表明,Ti0.9Cu0.1N@Pt/NCNTs催化剂的氧还原催化活性相对于TiMN@Pt具有更加明显的优势。

图3 TiNiN@Pt纳米粒子的高角环形暗场/扫描透射电子显微镜(HAADF/STEM)照片[70](a)和负载在碳纳米管上的TiCuN@Pt纳米粒子的HAADF/STEM照片及其氧还原过程示意图[2](b)Fig.3 HAADF/STEM images of TiNiN@Pt [70] (a) and TiCuN@Pt/NCNT [2] (b) nanoparticles
前过渡金属碳化物具有良好的电子导电性、耐腐蚀性和高熔点等特点[71],同时贵金属也极易在具有金属性质的前过渡金属碳化物表面与其形成强烈的相互作用,却不会形成稳定的碳化物[72]。因此,利用前过渡金属碳化物作为核,并在其表面沉积薄层贵金属制备具备核壳结构的催化剂,可很大程度上降低贵金属的使用量。Alba-Rubio团队[73]利用了这样的策略,制备出以WC为核、薄层Pt为壳层的WC@Pt核壳催化剂。首先他们将WOx与(NH4)2PtCl6混合均匀后,用SiO2在室温下进行包覆,得到SiO2/(NH4)2PtCl6/WOx复合物。接着在15% CH4/85% H2的气氛中,将上述得到的复合物进行多段温度热处理。当温度在200 ℃时,复合物中Pt从(NH4)2PtCl6中被还原出来,得到了SiO2/Pt/WOx复合物。温度升至600 ℃时,WOx被还原成W,在被SiO2包覆的中心位置与Pt形成Pt/W合金。而在900 ℃时,这些被包覆的Pt/W金属纳米簇因高温烧结形成单中心纳米粒子,此时由CH4分解而来的C与纳米粒子中的W形成WC。由于贵金属与WC不相溶,Pt从WC相中析出,并在其表面自组装形成均一的Pt壳层,得到SiO2包覆的WC@Pt。利用氢氟酸将SiO2刻蚀,即可得到WC@Pt催化剂。他们发现,以WC为核的低Pt核壳催化剂具有更好的电化学活性和稳定性,同时也具备更好的抗CO中毒的能力。通过理论计算表明,受WC核的影响,Pt与CO的吸附作用被弱化了,从而提高了其中Pt的抗CO中毒能力。
5 制备方法
核壳结构催化剂制备的难点在于,在作为基质的纳米粒子表面制备出单原子层或者仅有几个原子层厚度的超薄壳层,近年来发展起来的壳层制备技术主要包括:电化学沉积技术、表层去合金化以及化学还原法等。
5.1 电化学沉积法
制备核壳结构催化剂的电化学方法主要包括欠电位沉积法(underpotential deposition, UPD)和脉冲沉积法(pulse electrodeposition, PED)。
欠电位沉积,是指一种金属可在比其热力学可逆电位更正的电位下,以单层原子的形式沉积在另一金属上的现象。将欠电位沉积技术进一步应用于Pt基核壳催化剂的制备,即通过将金属表面已沉积的单层Cu原子层置换为Pt原子层从而形成薄层、完整的单原子层的Pt壳层(如图4所示)。利用欠电位沉积法制备的Pt基核壳结构催化剂,其制备过程通常在酸性介质中进行,而过渡金属在酸性介质中一般都不能稳定存在。因此,欠电位沉积法所选用的金属核通常为比Pt储量更大、更廉价的贵金属及其合金。此法虽仍采用了贵金属为核,成本上并未降低到以非贵金属为核的水平,但在保证高活性的同时很大程度上降低了Pt的使用,缓解了贵金属Pt价格昂贵和储量少等问题对燃料电池发展造成的困境。此方法适用于绝大部分以非Pt贵金属为核的催化剂体系,因此成为目前制备核壳结构催化剂的最常用的方法。

图4 欠电位沉积法制备核壳结构催化剂示意图Fig.4 The fabricating procedure of core-shell catalyst by underpotential deposition (UPD) method
脉冲电沉积法是指采用恒电流脉冲的方法,是作者课题组近年来提出和深入研究的一种制备核壳结构催化剂的方法,通过控制通导时间、关断时间来控制使合适量的Pt沉积于核上。通常事先将核及其复合材料涂布在脉冲电极上[37, 38],当电极导通时,电极极化增大,电极附近的Pt离子沉积于涂布在电极上的核粒子。当电极关断时,溶液本体中的Pt离子以迁移、对流和扩散等方式及时补充电极附近的Pt离子,并使其浓度恢复到正常值,完成一个脉冲沉积周期。通过调整脉冲周期次数,可有效控制核粒子表面的Pt沉积量,进而控制Pt层的厚度,条件合适时制得的催化剂的壳层厚度可达到2~4个原子层,且制得的催化剂的质量活性可媲美欠电位沉积技术制得的催化剂。与欠电位沉积法一样,脉冲沉积可大幅度减少贵金属Pt的使用,同时也具备超高的活性。不同的是,脉冲沉积不受限于核只能为金属,对其他导电的化合物也适用,近几年也有很多相关研究报道[70, 71]。因此,脉冲电沉积相比于欠电位沉积有更广泛的应用以及工业化潜力。
5.2 表面去合金化方法
表面去合金化是制备核壳结构催化剂的另一种十分重要的方法,通常是先制备Pt与其它金属M(M=Ni, Co, Fe等)合金的纳米卡结构(包括纳米粒子、纳米线、纳米管等),然后用酸等控制性地溶除表面层的M, 得到PtM@Pt的核壳结构催化剂粒子。按照近年来的相关报道,这种方法制备的催化剂通常都具有极高的活性。其不足是核中的M在质子交换膜燃料电池运行条件下容易溶出,致使燃料电池的性能急剧降低。美国阿贡国家实验室Stamenkovic教授团队[74]利用表面去合金化的方法制备出了高活性的PtNi@Pt核壳结构催化剂。他们将制备好的PtNi合金负载在碳粉上,通过在0.1 mol/L 的HClO4溶液中隔夜浸泡以洗去PtNi颗粒表面的Ni,然后在400 ℃下热处理以稳定PtNi表面的Pt,使其形成完整的Pt壳。
5.3 化学还原法
还原法是利用特定的表面活性剂以及还原剂控制金属前驱体的还原,使其形成核壳结构催化剂。常用的还原剂有NaBH4、水合肼、抗坏血酸、乙二醇等。此外,也可选择沸点较高的有机溶剂,在惰性气氛的保护下,用高温时易分解的金属前驱体,例如Pt(acac)2等还原制备核壳纳米粒子。还原法具有选择性强、对设备要求低、易操作等优点,具有大量制备Pt基核壳结构催化剂的潜力。其中较常见的一种化学还原法为胶体法。
胶体法是制备核壳结构催化剂常用方法之一。其主要步骤如下:先在溶液中形成非Pt胶体粒子,并以此为生长中心(种子),在聚电解质(如PVP等)保护剂存在的情况下,加入Pt化合物使其在胶体粒子表面还原包覆成壳。由于核种子表面保护剂的吸附,抑制了核粒子间的相互团聚,使其能稳定存在于溶液中,以便形成较完美的核壳结构催化剂粒子,如图5所示。胶体法操作简单,且催化剂形貌、粒子大小可以调控、分散性较好,但其所投入物料只能被部分还原利用,因此产出率很低,对原材料浪费较大。此外,该制备方法使用保护剂,可能最后得到的催化剂表面仍会吸附一些保护剂,这些保护剂有可能覆盖了催化剂的活性位,限制了催化剂的活性[26]。
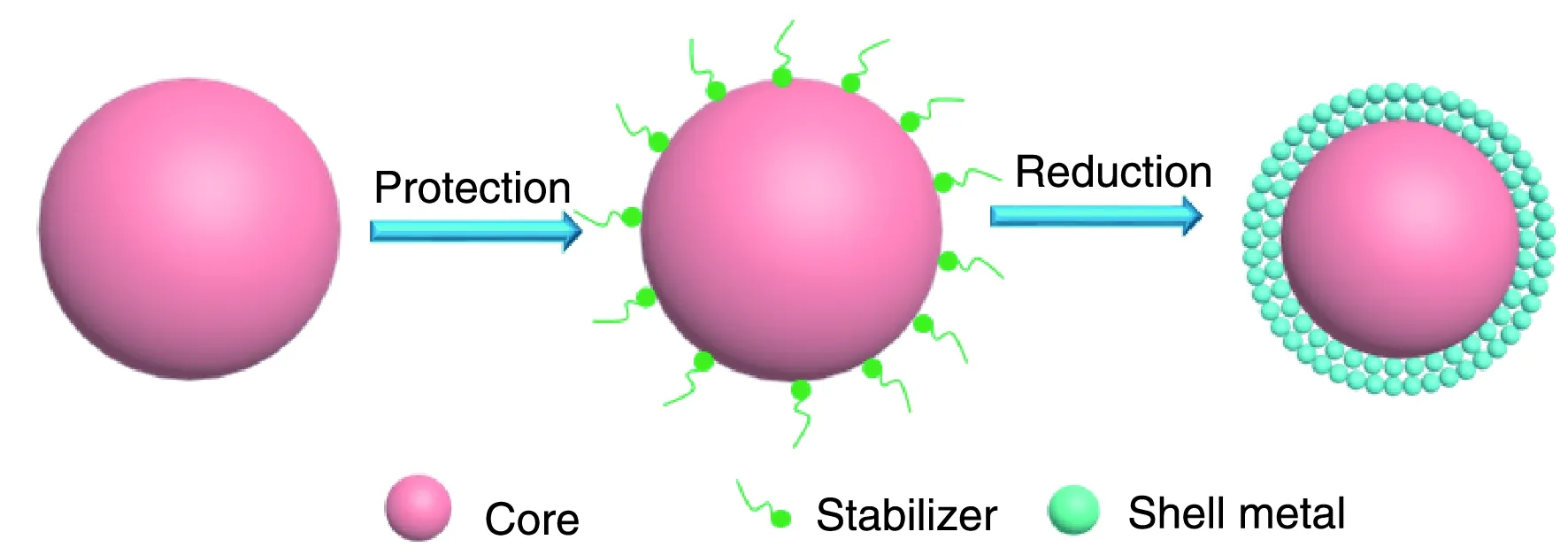
图5 胶体法制备核壳结构催化剂过程示意图Fig.5 The fabricating procedure of core-shell catalyst by colloid method
6 结 语
核壳结构低Pt催化剂可以大幅度提升Pt的质量活性,大幅度降低燃料电池的Pt的使用量,是降低燃料电池Pt使用量和燃料电池成本的希望之所在。近年来,核壳结构催化剂无论是在制备技术还是在新体系的研究方面均取得了长足的进步,但是,目前这类催化剂的商业化应用尚有许多难题需要攻克,如:核壳结构催化剂的放大制备问题、稳定性问题、贱金属组分的溶出问题等等。因此作者认为未来低Pt核壳结构催化剂研究应主要集中在以下几个方面:① 进一步优化合成核壳结构催化剂的合成工艺、制备条件,寻找一种稳定且可实现大规模生产的方法;② 当前Pd,Ir,Ru,Au等贵金属依旧是制备低Pt核壳结构催化剂核材料的首选,因此开发更廉价且能有效代替贵金属的新核材料,对大幅度降低Pt基核壳结构催化剂的制备成本、加快燃料电池商业化进程具有深远意义;③ 发展有效且清晰的催化剂材料表征方法及电化学测试方法可以从根源上探索催化剂作用机理、揭示核壳结构之间相互作用关系,为制备高性能的催化剂提供理论依据。
猜你喜欢
食品科学(2022年20期)2022-10-31
发光学报(2021年7期)2021-07-23
装备维修技术(2020年5期)2020-11-20
矿产综合利用(2020年1期)2020-07-24
中学生数理化(高中版.高二数学)(2020年2期)2020-04-21
分析化学(2018年1期)2018-01-18
车迷(2017年12期)2018-01-18
组织工程与重建外科杂志(2018年6期)2018-01-12
分析化学(2017年9期)2017-10-16
电子制作(2017年10期)2017-04-18
