
异相晶种诱导快速合成低硅菱沸石分子筛
2018-12-10 06:48李玉琴朱美华陈祥树
无机化学学报 2018年12期
李玉琴 胡 娜 张 飞 朱美华 吴 婷 陈祥树
(江西师范大学化学化工学院,先进材料研究院,分子筛膜材料国家地方联合工程实验室,南昌 330022)
近20年来,具有八元环骨架结构的小孔分子筛引起了研究者们的浓厚兴趣。这类分子筛的孔道大小与小分子气体和低碳烃类的分子动力学直径接近,它们被广泛应用于大分子/小分子、水/有机物或非极性分子/极性分子的分离[1-4]。具有典型三维八元环孔道结构体系的菱沸石分子筛是由CHA笼和双六元环(D6R)平行于c轴方向交替组成的骨架结构,其八元环孔道大小为0.38 nm×0.38 nm[5]。菱沸石分子筛以其高活性及择形性的特点在甲醇制低碳烯烃(MTO)的反应中具有广阔的应用前景[6-7]。同时,菱沸石分子筛的适中硅铝比(2~5)特性使得其表现出较强的亲水性和一定的耐酸性,是构筑高通量分子筛膜的理想膜材料[8-10]。
最初,菱沸石分子筛需借助有机模板剂来制备。Singh等[11]通过在前驱体溶胶中添加模板剂四甲基氢氧化铵,85℃下水热合成4 d得到硅铝比为2.4的菱沸石分子筛。昂贵有机模板剂的使用不仅大大提高了合成成本,而且会产生一定环境污染等问题。因此,国内外研究者们试图寻求一种绿色、高效、廉价的分子筛合成路线,即无模板剂法制备分子筛,这也是实现分子筛工业化制备的关键途径。无模板剂法制备菱沸石分子筛主要采用以下3种方式:晶型转化法[12-14]、原位合成法[15]和晶种诱导法[15-16]。Bourgogne等[12]首先仅仅将FAU型分子筛加入氢氧化钾溶液后制备出纯相的菱沸石分子筛。随后,Nouri等[13]采用相同制备方法,即以HY沸石分子筛为原料加入氢氧化钾碱溶液中,95℃下晶化96 h水热合成出菱沸石分子筛。Ridha等[14]同样成功利用晶型转化法在氢氧化钾溶液中加入Y型分子筛,95℃下晶化15 d合成出菱沸石分子筛。接着,Liu等[15]在不含有机模板剂和晶种的前驱体溶液中,160℃下晶化48 h原位合成出大颗粒的菱沸石分子筛(20~30 μm)。然而,以上 2种制备方法所需的合成时间都较长。因此,研究者们提出晶种诱导法来试图缩短菱沸石分子筛的合成时间。Li等[16]在不加有机模板剂的条件下,利用菱沸石分子筛晶种导向法,150℃下晶化2 d制备出菱沸石分子筛。Liu等[15]同样报道了在前驱体溶胶中加入0.5%(w/w)菱沸石分子筛晶种,其晶化时间从原位合成所需的48 h缩短至24 h。
晶种诱导法合成分子筛时,晶种可提供晶体有效的生长点或者生长面能快速诱导合成目标分子筛,这样极大地缩短了合成时间且可定向调控晶相,易于实现工业化生产。近年来,这种合成方法已广泛应用于 ZSM-34[17]、Beta[18]、LSX[19]、ZSM-5[20-21]、AlPO-17和SAPO-17[22]等分子筛的制备中。在我们前期的研究工作中报道了在无有机模板剂条件下借助异相的T型分子筛(具有结构相似的分子筛)可以诱导合成出低硅菱沸石分子筛[23],而从异相晶种到菱沸石分子筛晶体的生长机理缺乏深入地分析。同时,水热合成条件对菱沸石分子筛晶化过程的重要影响还需进一步探讨。因此,本文着重研究了晶种诱导合成菱沸石分子筛过程中的结晶规律,分析了晶种诱导作用,并揭示T型分子筛晶种诱导合成菱沸石分子筛的晶化机理。详细考察了合成条件如T型分子筛晶种添加量、n/n、n/n对分子筛合成的影响,并讨论合成液中Na+和K+对晶体生长的作用。
1 实验部分
1.1 菱沸石分子筛的合成
在不添加有机模板剂的条件下,通过异相晶种诱导合成出纯相的菱沸石分子筛。将硅溶胶(HS-40,质量分数为 40%,Sigma-Aldrich)、氢氧化钠(内蒙古红津化工有限公司,工业品)、氢氧化钾(上海青析化工科技有限公司,分析纯)、氢氧化铝(日本和光纯药试剂公司,分析纯)和超纯水(自制)按 n∶n∶n∶n=1∶(0.02~0.10)∶(0~0.8)∶(12~20)(M2O 为 Na2O 和K2O)配成300 g的反应液。初始溶胶在室温下搅拌老化12 h,再加入T型分子筛晶种搅拌均匀后装入不锈钢反应釜中在140℃下晶化0~24 h。T型分子筛晶种是根据文献报道的方法制备[24]。作为对比,老化完成后的初始溶胶直接装入不锈钢反应釜中在140℃下原位合成0~24 h。反应后,将所合成的分子筛用超纯水离心洗涤至中性,在100℃下烘干备用。
1.2 产物分析与表征
分子筛的形貌借助扫描电子显微镜(FE-SEM,Hitachi SU8020)和高分辨透射电子显微镜(TEM,JEOL JEM 2100)来观察。其中,SEM的加速电压为5 kV,所有样品均需喷铂金处理。TEM的加速电压为200 kV,样品溶于乙醇溶液,取上清液滴在铜网上干燥,无需喷金处理。用X射线衍射仪(XRD,Rigaku UltimaⅣ)表征所合成粉末的晶相。测试条件为采用Cu Kα辐射,波长为0.154 06 nm,管压为40 kV,管流为 40 mA,2θ扫描范围为 5°~45°,扫描速度为4°·min-1。固体产物的相对结晶度是以标准样品的 2θ分别为 9.4°、12.8°、20.5°、24.6°和 30.5°的5个菱沸石分子筛特征峰峰值之和作为参考值,其他样品与之相比计算得到。使用能量分散X射线分析(EDX,Bruker Quantax 200)对样品进行元素分析。样品测试前用导电胶固定在样品台上喷铂金处理,操作电压为20 kV。氮气吸附-脱附等温线采用美国麦克公司的ASAP 3000全自动物理吸附仪来测试。样品没有经过离子交换处理。测试时,样品在350℃的真空环境中预处理6 h,在-196℃下用液氮与其充分接触达到吸附平衡。根据氮气的加入量和吸附平衡后的残余量,计算氮气的吸附量。粉末样品的27Al MASNMR共振频率为104.2 MHz,所使用的Bruker AdvanceⅢ400 WB核磁共振谱仪中氧化锆转子的直径为4 mm。在干燥空气中,27Al MASNMR的转子旋转为15 kHz。27Al MAS NMR测定累计3μs的脉冲,2 s的循环延迟和1 024次扫描。Al(NO3)3·9H2O 用 作27Al MAS NMR 的 化 学 位 移 参考。 通过紫外拉曼光谱仪(UV-Raman)(DL-2)来进行样品的骨架结构分析,采用325 nm激光光源。
2 结果与讨论
2.1 低硅菱沸石分子筛晶化过程
图1为原位合成和添加0.5%(w/w)T型分子筛晶种在不同晶化时间下合成分子筛的XRD图。表1列出了不同合成条件下所得的分子筛情况。这些分子筛的合成溶胶中(n+n)/n为0.38。从图1a可以看到,合成溶胶在无晶种诱导的作用下晶化2 h呈无定形相 (样品P1)。随着晶化时间延长,2θ=5.55°、11.77°、19.30°、22.66°、28.00°、30.71°等处开始出现强峰,与标准谱图对比发现该晶相归属于L型分子筛的特征峰[25]。当晶化时间延长至24 h(样品P6),这些峰的强度大大加强,未出现其他杂峰。这表明无晶种诱导下溶胶原位合成出纯相L型分子筛(样品 P2~P6)。 当溶胶中加入 0.5%(w/w)T 型分子筛时,未进行晶化的合成溶胶呈无定形状态(图1b,样品P7),说明T型分子筛加入合成溶胶后发生溶解。当晶化时间为 2 h 时 (样品 P8),2θ=9.40°、12.79°、20.45°、24.61°、30.45°、30.73°等处开始出现强峰,与标准谱图对比发现该晶相归属于菱沸石分子筛的特征峰[8-9,25]。这表明加入的晶种溶解在晶化2 h内起着关键的作用,即消除诱导期直接开始形成菱沸石晶相,大大缩短了合成时间。随着晶化时间增加,这些特征峰强度呈现先增加后基本保持不变的趋势,未出现其他杂峰。这些结果显示菱沸石晶相在晶化时间为2 h时快速生成,即使延长晶化时间至24 h,仍可获得纯相菱沸石晶体(样品 P8~P13)。根据图1b中不同晶化时间下晶体强度关系结果得出其晶化曲线图(图2)。从晶化曲线图可以看出,菱沸石晶体在晶化时间为0~8 h之间晶体快速生长,当晶化时间为8 h时(样品P10),晶体达到稳定期,相对结晶度达到最高。此时整个体系达到晶化平衡,即晶体生长速率与晶体溶解速率平衡状态。而当继续延长晶化时间导致结晶度略有下降,这与分子筛是介稳相有关。图3为不同晶化条件下分子筛的SEM图。当添加0.5%(w/w)T型分子筛晶化时间为2 h时(样品P8),出现由许多小晶粒组成的核桃状菱沸石晶体形貌,但这些小晶粒未生长完全(图3b)。当晶化时间为6 h时(样品P9),核桃状菱沸石晶体上的小晶粒呈现较好生长,小晶粒变大且形貌更清晰 (图3c)。当最佳晶化时间为8 h时(样品P10),小晶粒生长完全,菱沸石分子筛呈现出典型核桃状的特征形貌,晶体上小晶粒大小均匀,晶体颗粒大小约为1.5 μm(图3d)。这些表征结果与图2中8 h时分子筛结晶度最高的结论相吻合。而当未添加晶种晶化2 h时,样品P1的形貌为无定形状(图3e)。当原位晶化时间延长至6 h时(样品P2),仅合成出大小不均一的小块状L型分子筛晶体形貌(图3f)。结合表1中菱沸石分子筛P8~P13的晶体性质来看,晶体的硅铝比未随晶化时间变化而发生明显的变化,仅结晶度随之增强直至到达平衡状态。与传统晶型转化法合成菱沸石分子筛[23](晶种量与溶胶量的比值为10%(w/w);合成时间为 96 h)相比,采用 T 型分子筛诱导合成的菱沸石分子筛不仅添加的晶种量 (晶种量与溶胶量的比值为0.5%(w/w);合成时间为8 h)是前者的1/20,合成时间也仅为前者的1/12。

图1 (a)原位合成和(b)添加0.5%(w/w)T型分子筛时在不同晶化时间下合成分子筛XRD图Fig.1 XRD patterns of zeolites synthesized by (a)in-situ and (b)adding 0.5% (w/w)zeolite T with different crystallization times
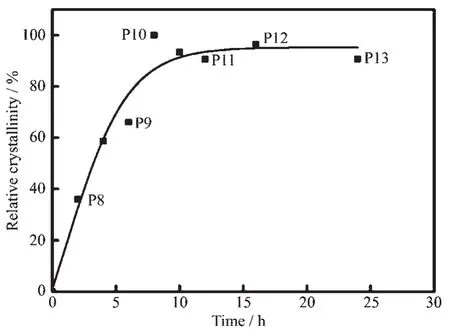
图2 添加0.5%(w/w)T型分子筛时合成的菱沸石分子筛晶化曲线图Fig.2 Crystallization curves of chabazite zeolites synthesized by adding 0.5%(w/w)zeolite T
图4 a为晶种诱导合成的低硅菱沸石分子筛(P10)的氮气吸附-脱附等温线。由图可知,分子筛的氮气吸附量不高,吸附分支变化缓慢,氮气脱附时与吸附的等温线不重合,脱附分支在中等大小相对压(P/P0=0.5~0.9)处形成了滞后环,对应与典型的IUPAC分类中Ⅳ型,存在介孔特性,可能分子筛中存在二次堆积孔,这说明分子筛具有多级孔结构[26]。根据我们前期的研究工作[23],P10氮气吸附量不高可能是合成的分子筛骨架含铝量高,导致大量钠离子在孔道内的占位效应而导致堵孔,或者是因为合成的含钠分子筛骨架中存在大量的堆垛层错现象[27]。图4b为低硅菱沸石分子筛(P10)的27Al MAS NMR谱图。从谱图可以看出,仅在化学位移δ=59出现一个强峰,归属于分子筛骨架中铝四配位的峰,证实不存在骨架外铝物质。

表1 不同合成条件下所得产物的性质Table 1 Properties of products obtained under different synthesis conditions

图3 (a)T 型分子筛和不同晶化条件下制备的(b)P8,(c)P9,(d)P10,(e)P1 和(f)P2 分子筛的 SEM 图Fig.3 SEM images of(a)zeolite T and zeolites synthesized with different crystallization times of(b)P8,(c)P9,(d)P10,(e)P1 and (f)P2

图 4 低硅菱沸石分子筛(P10)的(a)氮气吸附-脱附等温线和(b)27Al MASNMR 谱图Fig.4 (a)N2 adsorption-desorption isotherms and (b)27Al MASNMR spectra for low-silica chabazite crystal(P10)
2.2 晶种量的影响
在添加晶种诱导过程中,晶种量与晶体生长速率成正比关系[28]。图5给出了添加不同含量的T型分子筛作为晶种合成分子筛的XRD图。未添加晶种条件下只合成出L型分子筛(样品P3),随着晶种量的增加,晶体结晶度增强,这表明晶体生长速率加快。当添加晶种量为 0.5%(w/w)时(样品 P10),分子筛显示出纯相的菱沸石分子筛特征峰。当加入晶种量为 1.0%(w/w)时(样品 P14),XRD 图中出现了 T 型分子筛的特征峰,这可能是因为T型分子筛在初始晶化时含量过高,部分晶种来不及溶解,导致物相中产生菱沸石和T型分子筛共生的现象。图6给出的是添加T型分子筛晶种量分别为0%、0.5%和1.0%(w/w)时合成分子筛的SEM图。未添加晶种合成的L型分子筛(样品P3)为纳米级的正方体晶体形貌(图 6a)。 当添加最佳晶种量为 0.5%(w/w)时(样品P10),分子筛为典型的核桃状菱沸石特征形貌 (图6b)。当加入晶种量为 1.0%(w/w)时(样品 P14),分子筛为核桃状菱沸石晶体和棒状T型分子筛晶体的交错生长(图6c)。这些结果与XRD表征结果一致。
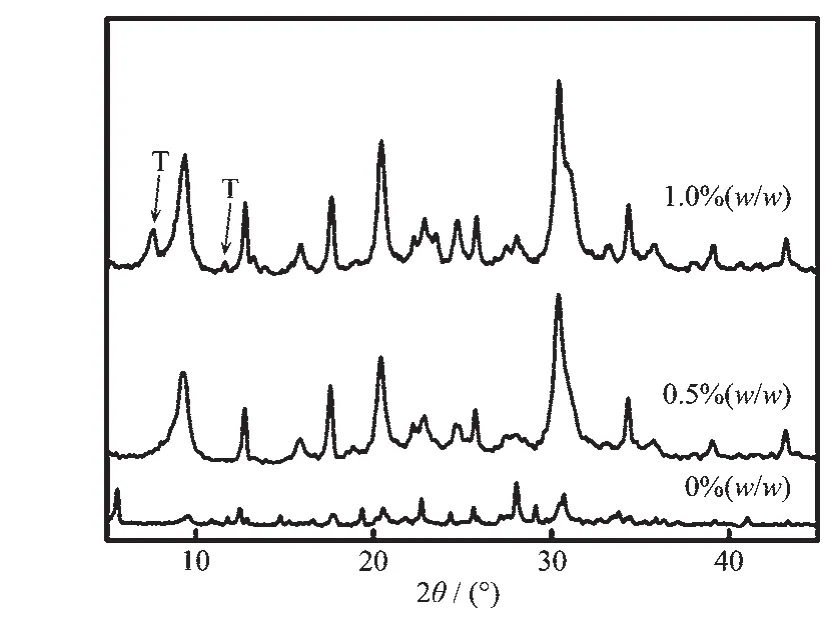
图5 添加不同含量T型分子筛时合成分子筛的XRD图Fig.5 XRD patterns of zeolites synthesized by adding different seed contents of zeolite T

图6 添加不同含量T型分子筛时合成分子筛的SEM图Fig.6 SEM images of zeolites synthesized by adding different seed contents of zeolite T
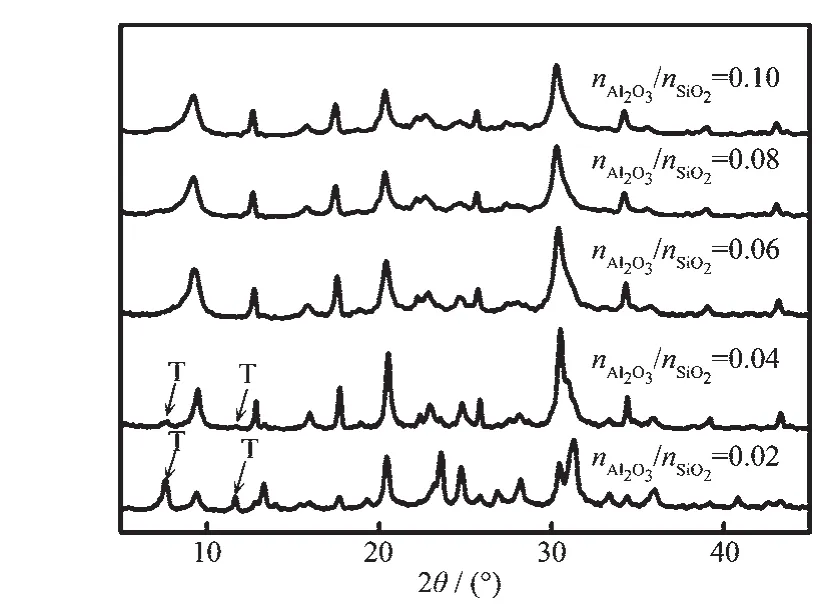
图7 不同n/n下合成分子筛的XRD图Fig.7 XRD patterns of zeolites synthesized with different n Al2 O3/n SiO2ratios

图8 不同n/n下合成分子筛的SEM图Fig.8 SEM images of zeolites synthesized with different/nratios
2.4 n H2 O/n SiO2的影响
图9 给出了添加0.5%(w/w)T型分子筛作为晶种的溶胶中,不同n/n下合成分子筛的XRD图。从XRD图可以看到,当溶胶n/n为20时(样品P20),合成溶胶浓度太稀,无法提供足够“营养”成核,导致晶体无法生长,表现为无定形状态。随着n/n逐渐降低,合成溶胶浓度增加,晶体生长速率加快。当n/n为16时(样品P10),形成了纯相的菱沸石分子筛。当n/n降低至12时(样品P19),合成液浓度过稠,粘度变大,最终产物为菱沸石和T型分子筛的混晶。图10为不同n/n下合成分子筛的SEM图。n/n为12时所合成的分子筛(样品P19)中出现T型分子筛的共生生长(图10a)。 最佳 n/n为16时所合成的分子筛 (样品P10)表现为纯的核桃状菱沸石晶体(图10b)。当nH2O/n增大至20时(样品P20),只出现无定形的形貌(图 10c)。

图9 不同n/n下合成分子筛的XRD图Fig.9 XRD patterns of zeolites synthesized with different n/nratios

图10 不同n/n下合成分子筛的SEM图Fig.10 SEM images of zeolites synthesized with different n/nradios
2.5 Na+和 K+的影响
在无有机模板剂合成过程中,金属阳离子通常起着结构导向和调节pH值等重要作用[29]。本文中低硅菱沸石分子筛是采用Na+和K+两种金属离子共同作用的硅铝体系来合成,考察Na+和K+对分子筛晶化过程的作用尤为重要。图11给出了溶胶中不同n/n和n/n体系下对应合成的分子筛晶相情况。这些分子筛的合成条件为:溶胶的物质的量配比为16,晶化温度为140℃,晶化时间8 h,添加晶种量为 0.5%(w/w)。 当溶胶中 n/n较高 (0.285或0.380)时,n/n在较大范围内变化均可合成出高结晶度的菱沸石分子筛。尤其当n/n为0.285时,n/n从0.095至0.285范围内可合成出纯菱沸石晶相。当溶胶中n/n较低时,即使n/n不断增大只能形成L型分子筛。所以,合成溶胶中(nNa2O+nK2O)/nSiO2的最佳比值为 0.38。已有文献报道无有机模板剂合成FER型分子筛时,K+对形成FER的基本结构单元起主要作用,而且K+在晶化过程中能有序排列已形成的基本结构单元。在Na+的合成体系中引入K+有利于促进组织四元环形成双六元环(D6R)结构,从而形成目标分子筛[30]。菱沸石分子筛骨架结构是由CHA笼和D6R组成,它们的次级结构单元为四元环。因此,在本研究中,Na+在无定形溶液中起着形成四元环和有序组装基本结构单元的作用,而K+只对组织四元环形成基本结构单元的过程起着促进作用,这个结论与Imai等[31]一致。

图11 不同n/n和n/n下合成的分子筛晶相关系图Fig.11 Crystal phase diagrams of zeolites synthesized with different n/nand n/nratios
2.6 T型分子筛晶种诱导合成菱沸石分子筛的晶化机理
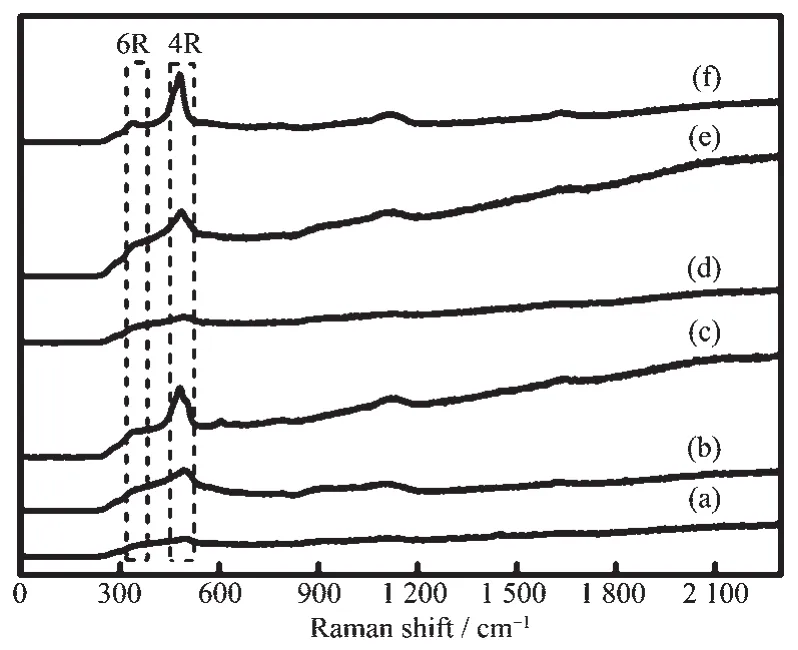
图 12 原位晶化(a)0,(b)2,(c)4 h 和添加 0.5%(w/w)T 型分子筛晶化(d)0,(e)1,(f)1.5 h 合成分子筛的紫外拉曼图Fig.12 UV Raman spectra for zeolites in-situ crystallized with (a)0,(b)2,(c)4 h and crystallized with (d)0,(e)1 and (f)1.5 h by adding 0.5% (w/w)zeolite T
图12 给出了不同晶化时间下未添加和添加T型分子筛作为晶种所合成分子筛的紫外拉曼图。这些分子筛的合成条件为:溶胶的物质的量配比为nSiO2∶nAl2O3∶(nNa2O+nK2O)∶nH2O=1∶0.06∶0.38∶16,晶化温度为140℃,添加晶种量为0.5%(w/w)。当晶化时间为0 h时,添加和未添加晶种的溶胶紫外拉曼图相似,均在500 cm-1左右出现宽的不对称峰,归属为无定形溶胶中四元环的振动峰[32-33](样品P7)。添加T型分子筛晶种诱导晶化1 h时,分别在480和350 cm-1出现了明显的四元环和六元环的振动峰[32-33],这些次级结构单元相互连接构成菱沸石分子筛的骨架结构。未添加T型分子筛晶种的条件下,晶化时间延长至4 h,分别在320 cm-1出现微弱的六元环和498 cm-1出现四元环的振动峰,同时在601 cm-1出现弱峰,其归属为L型分子筛的特征峰[34]。这些结果与图1中XRD表征结果一致。以上结果表明,在分子筛合成初始阶段,无定形合成液的次级结构单元主要是四元环,T型分子筛的加入未改变合成液的次级结构单元,这一结论与已报道的文献结果相似[35-36]。ERI骨架为主的T型分子筛晶体中二次结构单元四元环被认为是诱导形成菱沸石分子筛特征笼(CHA笼)的关键因素。在一定的水热条件下,T型分子筛晶体不断溶解释放出六元环和四元环,这些结构单元能迅速成为菱沸石分子筛的特征结构单元,抑制L型分子筛的特征单元和特征笼(不含四元环的CAN笼)的形成。
图13为不同晶化时间下合成分子筛的TEM图。合成溶胶未添加T型分子筛时,呈现无定形状态(图 13a)。 当原位晶化 2 h 时(样品 P1),晶格条纹大小约为2 nm的细小颗粒出现在无定形溶胶内(图13b)。相应的XRD表征并未检测出这些小颗粒的存在(图1a)。当原位晶化时间延长至6 h时(样品P2),这些小颗粒通过不断消耗无定形溶胶中的“营养物质”尺寸变大,逐渐开始形成晶体(图13c)。结合XRD、SEM和紫外拉曼等表征结果,我们认为原位合成L型分子筛的晶化机理为:最初无定形溶胶中的硅铝酸盐在高碱水热条件下形成以四元环为主的次级结构单元。随着合成时间的延长,六元环的次级结构单元数量逐渐增多,利于L型分子筛(不含四元环的CAN笼)骨架结构的形成进而产生晶核。通过不断消耗溶胶中“营养物质”,晶核外延生长并不断晶化形成L型分子筛晶体。由图13d可见,当合成溶胶中添加 0.5%(w/w)T 型分子筛时(样品 P7),无定形溶胶内出现许多球状颗粒,这些颗粒显示出明显的晶格条纹,大小约为5 nm。这说明晶种接触溶胶后发生溶解,并被无定形溶胶包裹,其相应的XRD图为无定形状态 (图1b)。随着晶化时间的延长,晶体尺寸逐渐增大(图13e和13f)。当晶化时间为2 h时(样品P8),球状晶体数量增多且尺寸增大至约15 nm,溶胶中无定形状态逐渐消失,开始形成固体晶体且结晶度逐渐增强(图13f),与图1b中出现弱的菱沸石分子筛衍射峰相吻合。这可能是因为包裹在无定形溶胶中的胶团粒子提供了目标分子筛生长所需的更多晶化面,当晶化时间增加时,胶团粒子本身通过合成液的溶胶原位重排机制转变成菱沸石晶格骨架。

图 13 原位晶化(a)0,(b)2,(c)6 h 和添加 0.5%(w/w)T 型分子筛晶化(d)0 h,(e)1.5 h,(f)2 h 合成分子筛的 TEM 图Fig.13 TEM images for zeolites in-situ crystallized with (a)0 h,(b)2 h,(c)6 h and crystallized with (d)0 h,(e)1.5 h and (f)2 h by adding 0.5% (w/w)zeolite T
初始合成溶胶呈粘稠状,通过加热晶化,逐渐溶解形成具有前驱体的无定形硅铝活性溶胶。当加入T型分子筛晶种后,随着合成时间的延长,菱沸石分子筛晶体随之生长,无定形硅铝“营养液”逐渐减少直至消失,而T型分子筛晶种的加入没有改变溶胶中次级结构单元。这说明晶种诱导合成分子筛晶化过程异于传统的“核壳生长机理”。类似的诱导晶化行为也发生在MTW[35]和β[36]等分子筛制备中。基于上述一系列表征结果,我们认为在含钠钾的硅铝酸盐溶胶中加入异相晶种(T型分子筛)诱导快速形成菱沸石分子筛的晶化机理如图14所示。在初始溶胶中加入诱导分子筛后,异相晶种不断溶解并解聚形成纳米粒子被无定形溶胶包裹,随着无定形溶胶的逐渐溶解重组形成具有前驱体的活性溶胶;此时嵌入在无定形溶胶内的胶团粒子与活性溶胶一接触,提供了利于菱沸石分子筛形成的活性晶面,迅速形成菱沸石分子筛的晶格骨架并逐渐生长成为目标分子筛。
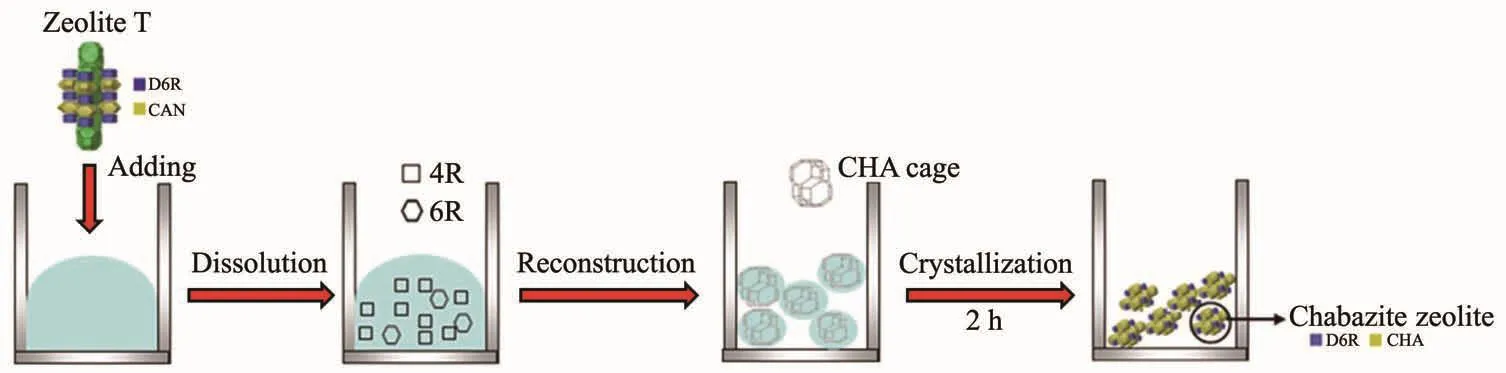
图14 异相晶种诱导合成菱沸石分子筛的晶化机理示意图Fig.14 Schematic diagram of crystallization mechanism of chabazite zeolites induced by heterogenous seeds
3 结 论
在含钠钾双离子的合成溶胶中,通过加入异相晶种诱导快速制备出纯相的低硅菱沸石分子筛。相同合成条件下未添加晶种时自成核形成L型分子筛,而加入T型分子筛晶种诱导晶化2 h得到菱沸石分子筛。其优化的合成条件为:晶化时间为8 h,添加晶种量为 0.5%(w/w),n/n为 0.06,n/n为 16 和(n+n)/n为 0.38。Na+在无定形溶胶中有利于四元环的形成和基本结构单元的有序组装,而K+在组织四元环形成基本结构单元的过程起着促进作用。结合各种表征分析,揭示了T型分子筛异相晶种诱导合成菱沸石分子筛的晶化机理。即T型分子筛加入初始溶胶后迅速溶解并解聚形成纳米粒子被无定形溶胶包裹。随后,这些胶团粒子与无定形溶胶逐渐溶解并重组形成具有前驱体的活性溶液一接触,利于形成菱沸石分子筛的活性晶面,并迅速形成菱沸石分子筛的晶格骨架,逐渐生长成纯相的低硅菱沸石分子筛。
猜你喜欢
化学工业与工程(2022年4期)2022-09-20
石油炼制与化工(2022年9期)2022-09-05
四川化工(2021年6期)2022-01-12
铁道标准设计(2021年9期)2021-09-26
中国材料进展(2020年4期)2020-05-23
应用化工(2019年10期)2019-11-05
火工品(2019年3期)2019-09-02
制造技术与机床(2019年2期)2019-03-06
西北药学杂志(2018年5期)2018-09-20
湖北农业科学(2016年5期)2016-10-19
