
苏云金芽胞杆菌aiiA的5'端侧翼序列的克隆与功能鉴定
2018-11-30 03:09:00陈少威吴程苏月华蔡斌斌谢盼盼杨梅
生物技术通报 2018年11期
陈少威 吴程 苏月华 蔡斌斌 谢盼盼 杨梅
(福建师范大学生命科学学院,福州 350117)
大多数植物病原体是革兰氏阴性细菌[1],其自身代谢产物N-酰基高丝氨酸内酯(N-Acylhomoserine lactones,AHLs)已被证明是参与群体感应(Quorum sensing,QS)系统中关键的信号分子。细菌能通过QS系统感知菌群密度并调节其生理活动[2],当菌群中AHLs浓度达到一定阈值后,就能激活病原菌致病基因表达,使动植物出现致病性,如参与病原体和植物之间相互作用的毒力因子[3-4]。因此,科学家提出了一种植物病原体控制策略,即破坏病原体的QS系统可能下调菌群密度,从而抑制可能感染宿主植物的毒力因子的表达。
当前对N-酰基高丝氨酸内酯酶(N-acylhomoserine Lactonase,AiiA蛋白)编码基因aiiA的研究大部分侧重于对其进行克隆表达。苏云金芽胞杆菌aiiA编码的AiiA蛋白,通过水解内酯键失活AHLs,从而破坏AHLs介导的QS系统[5-6]。有研究表明,在微生物中,蜡状芽孢杆菌AiiA蛋白对魔芋软腐病菌 CZY 表现出较强的抗病活性[7];Álvaro等[8]分离到一株苏云金芽胞杆菌,克隆其aiiA并转入大肠杆菌,发现重组的大肠杆菌在含有欧文氏杆菌的培养基中共培养时,可以干扰AHLs信号的积累或响应,破坏其QS系统,这与Wu等[9]从苏云金芽胞杆菌中克隆到的aiiA在毕赤酵母GS115中表达,其提取物表现出对欧文氏杆菌的抗菌活性有相似之处。同时,有学者将aiiA转化到巨尾桉中后,用青枯雷尔氏菌侵染植株后,发现转基因的巨尾桉出现萎蔫现象的时间显著延迟,发病率降低,表明转入aiiA能增强其抗青枯病能力[10]。同样,将在巴斯德毕赤酵母中异源表达的AiiA蛋白与嗜水气单胞菌的同时注射进鲤鱼时,其死亡率与单独注射嗜水气单胞菌相比,降低了约25%[11]。因此,使用AiiA蛋白破坏QS系统被认为是生物防治病害的有效手段。然而,aiiA在苏云金芽胞杆菌中的表达非常低,对aiiA启动子的转录调控研究很少。因此,研究aiiA的转录机制可能是增强AiiA蛋白表达量的有效方法,启动子区域的寻找是研究转录机制的第一步。
本研究通过克隆aiiA的5'侧翼区域,利用在线生物软件分析潜在的启动子区域。以gfp作为报告基因,通过分段克隆及定点突变,对aiiA的5'端侧翼序列的启动子功能进行研究。分析苏云金芽胞杆菌aiiA启动子的转录调控功能,为aiiA在不同宿主中的高表达提供理论基础,也为细菌病害的生物防治提供新的思路。
1 材料与方法
1.1 材料
大肠杆菌E.coliDH5α、E.coliBL21(DE3)由福建师范大学生命科学学院工程研究中心保存,苏云金芽胞杆菌BRC-ZLL5(Bacillus thuringiensis)由福建农林大学生命科学学院关雄教授惠赠。苏云金芽胞杆菌aiiA由福建师范大学生命科学学院工程研究中心前期克隆得到。所有菌株均用LB培养基培养。绿色荧光蛋白表达质粒pET28a-gfp以及由pET28agfp质粒敲除T7启动子改造而来的绿色荧光蛋白本底表达质粒pET28a-dP-gfp由福建师范大学发育生物学重点实验室保存,pMD18-T载体购自TaKaRa公司。
1.2 方法
1.2.1 苏云金芽胞杆菌aiiA的5'端侧翼序列的克隆 将克隆的aiiA(GenBank登录号:DQ440581.1)在NCBI网站进行BLAST检索,选择同源性最高的苏云金芽胞杆菌全基因组序列(GenBank登录号:CP001903.1),在aiiA起始密码子ATG上游1 900 bp处设计特异性PCR扩增引物,上游引物P1插入BglⅡ 酶切位点:5'-GAAGATCTAGC TAGTGTAAAATGACGTTTGCTT-3';下游引物P2插入NheⅠ酶切位点:5'-CTAGCTAGCTATGTA TCCACCTTTACATTTAGTT-3'(下划线处碱基为限制性内切酶识别位点)。此外,在aiiA起始密码子ATG下游200 bp处设计另一条下游引物P3:5'-GTACCGTTAAAAAGCCCTTCATTAT-3',可以克隆出一条既包含aiiA的5'端侧翼序列,又包含200 bpaiiA的目的序列。本研究引物均由上海生工生物工程股份有限公司合成。
为扩增aiiA的5'端侧翼,使用分子克隆方法提取苏云金芽胞杆菌的质粒[12],进行PCR扩增,扩增体系为模板 DNA 1 μL、dNTP 0.5 μL、10×buffer 1.5 μL、引物各 0.5μL、ExTaq 酶 0.15 μL 和 ddH2O 10.9 μL。反应条件为 94℃ 5 min ;94℃ 30 s,55℃ 30s,72℃ 2 min,30个循环;72℃ 10 min。PCR扩增产物经1%琼脂糖凝胶电泳后纯化回收、连接和转化。筛选阳性克隆测序。插入aiiA的5'端侧翼区的质粒pMD18-T命名为pMD18-T-aP-1930-+200。
1.2.2 苏云金芽胞杆菌aiiA的5'端侧翼序列aP-1930-+200启动子区域分析 使用在线软件BDGP Neural Network Promoter Prediction,V2.2(http://www.fruitfly.org/seq_tools/promoter.html)、Promoter 2.0Prediction Server(http://www.cbs.dtu.dk/services/Promoter/)和NCBI 中Finding Promoter数据库预测aiiA的5'端侧翼序列可能的启动子区域。
1.2.3 苏云金芽胞杆菌aiiA的5'端侧翼序列的分段克隆 以克隆的侧翼序列aP-1930-+200为模板,根据启动子潜在区域预测结果,在aiiA的5'端侧翼序列的不同位置设计10对引物(表1),进行分段克隆。上游引物加入BglⅡ酶切位点,下游引物加入NheⅠ酶切位点。

表1 aiiA的克隆引物
以pMD18-T-aP-1930-+200质粒为模板进行PCR扩增。扩增体系同1.2.1。其中片段aP-100--1、aP-295--1、aP-429--1、aP-429--296和aP-295--101的 PCR 反应条件为94℃ 5 min,94℃ 30 s,55℃ 30 s,72℃ 30 s,30 个循 环 ;72℃ 10 min;片 段aP-531--1和aP-1930--1430的PCR 反应条件为94℃ 5 min;94℃ 30 s,55℃ 30 s,72℃ 1 min,30个循环 ;72℃ 10 min;片段aP-1031--1、aP-1531--1和aP-1931-1的PCR反应条件为94℃ 5 min;94℃ 30 s,55℃ 30 s,72℃ 2 min,30 个循环 ;72℃10 min。
1.2.4 启动子功能鉴定载体pET28a-aP-x-gfp的构建 将aiiA的5'端侧翼序列的分段克隆与经BglⅡ和NheⅠ双酶切的pET28a-gfp质粒用T4 DNA连接酶16℃ 过夜连接,然后将连接的pET28a-aP-x-gfp载体转化到大肠杆菌E.coliBL21(DE3)感受态细胞,并命名为E.coliBL21(DE3)-pET28a-aP-x-gfp。
1.2.5aiiA的5'端侧翼区域的启动子活性分析 将重组大肠杆菌E.coliBL21(DE3)-pET28a-aP-x-gfp在50 mL含有50 μg/mL卡那霉素的LB培养基中37℃震荡孵育过夜。当细菌浓度达到OD600=0.6-0.8时,向培养基中加入IPTG(终浓度1.0 mmol/L)。取5 μL菌液制成玻片标本后,直接用荧光显微镜观察(激发光为蓝光),通过荧光强度比较启动子的活性。大肠杆菌E.coliBL21(DE3)-pET28a-gfp、E.coliBL21(DE3)-pET28a-dP-gfp分别作为阳性对照和阴性对照。取3.5 mL上述诱导过的菌液于荧光比色皿中,利用荧光分光光度计在激发光波长(Ex)为491 nm、发色光波长(Em)为509 nm下测量菌液荧光强度。
1.2.6 苏云金芽胞杆菌aiiA基因启动子的定点突变 在载体pET28a-aP-295--101-gfp基础上,对aiiA启动子-150--142 bp的2个TATA序列进行点突变,结合待突变位点设计4对引物(表2):前2对为-144和-147位点处的单点突变引物,第3对为-144位和-147位点处的双点突变引物,第4对为2个TATA序列缺失突变引物。

表2 突变引物
定点突变PCR扩增体系为模板DNA 0.4 μL、dNTP Mixture 1.6 μL、5×Prime STAR buffer 4.0 μL、引物各 0.4 μL、Prime STAR HS DNA Polymerase 0.2 μL和ddH2O 13.0 μL。PCR反应条件为94℃ 5 min;94℃ 30 s,51℃ 30 s,72℃ 7 min,30 个循环 ;72℃10 min。PCR扩增产物经琼脂糖凝胶电泳验证正确后,冰浴2-3 min,向反应物中加入1 μLDpnⅠ酶,消化模板质粒pET28a-aP-295--101-gfp,37℃ 水浴过夜后热击转化大肠杆菌E.coliBL21(DE3)感受态细胞。
通过DNA测序鉴定阳性诱变菌株,通过荧光显微镜和荧光分光光度法分析含有突变的5'侧翼突变区域的重组菌株。
2 结果
2.1 aiiA的5'端侧翼序列的克隆与分析
以苏云金芽胞杆菌质粒DNA为模板,用引物P1和P2、P1和P3进行扩增,获得约1.9和2.1 kb大小片段。将2.1 kb片段测序结果进行在线BLAST(http://www.ncbi.nlm.nih.gov),结果显示克隆的aiiA的5’端侧翼序列与已经公布的苏云金芽胞杆菌YBT-1520(GenBank登 录 号:CP007607.1)aiiA的5’端侧翼序列同源性为100%,其中3’端的200 bp与aiiA的5’端同源性为100%(GenBank登录号:DQ000642.1)。所克隆的片段在GenBank登录号为KP690195。
利用在线分析软件BDGP Neural Network Promoter Prediction对aiiA的5’端侧翼序列进行分析,发现其含有潜在的启动子区域(图1红色区域),其中位于-1 395--1 346 bp和-339--292 bp处的启动子区域可靠性最高,分值均为满分1.0;通过NCBI中Finding Promoter数据库预测,发现其可能存在2个启动子区域(图1下划线区域),分别位于-429--1 bp和-1 930--1 822 bp处。此外,在序列-150--142 bp处还存在2个连续的TATA序列,可能对启动子活性有很重要的作用。
2.2 aiiA的5'端侧翼序列的启动子活性鉴定
以pMD18-T-aP-1930-+200质粒为模板,进行PCR扩增。琼脂糖凝胶电泳(图2)可知在100、295、134、429、195、501、531、1 031、1 531 和 1 930 bp处出现目的条带,PCR扩增产物与目的片段大小一致,产物特异性好。
由模板质粒pET28a-dP-gfp构建而来的分段克隆载 体 pET28a-aP-x-gfp(pET28a-aP-100--1-gfp、pET28aaP-295--1-gfp、pET28a-aP-295--101-gfp、pET28a-aP-429--1-gfp、pET28a-aP-429--296-gfp、pET28a-aP-531--1-gfp、pET28aaP-1031--1-gfp、pET28a-aP-1531--1-gfp、pET28a-aP-1930--1-gfp和pET28a-aP-1930--1429-gfp)经过双酶切(BglⅡ和NdeⅠ)和DNA测序鉴定,10个克隆子均显示了正确的条带,构建的载体经上海生工生物股份有限公司序列测定后表明插入序列与目的序列相同,表明载体 pET28a-aP-295--101-gfp、pET28a-aP-100--1-gfp、pET28a-aP-429--296-gfp、pET28a-aP-429--1-gfp、pET28aaP-295--1-gfp、pET28a-aP-531--1-gfp、pET28a-aP-1531--1-gfp、pET28a-aP-1031--1-gfp、pET28a-aP-1930--1429-gfp和pET28a-aP-1930--1-gfp构建成功。
培养含有pET28a-aP-x-gfp载体的重组大肠杆菌菌株,使用荧光显微镜和荧光分光光度计分析启动子活性。荧光显微镜(图3)显示大肠杆菌菌株BL21(DE3)-pET28a-aP-1930--1429-gfp,BL21(DE3)-pET28a-aP-295--1-gfp和BL21(DE3)-pET28a-aP-295--101-gfp能够产生绿色荧光,表明aiiA的5'侧翼区域aP-1930中的aP-1930-1429、aP-295--1和aP-295--101具有启动子活性,其中aP-1930--1429和aP-295--101区域荧光强度最高。
荧光分光光度法进行定量分析(图4)显示BL21(DE3)-pET28a-aP-1930--1429-gfp菌株启动子活性最高,其次是BL21(DE3)-pET28a-aP-295--101-gfp菌株,BL21(DE3)-pET28a-aP-295--1-gfp菌株启动子活性相对较低。
2.3 定点突变对苏云金芽胞菌aiiA的5'端侧翼区域启动子活性的影响
以pET28a-aP-295--101-gfp质粒为模板进行定点突变,测序结果显示缺失突变质粒序列TATA盒被成功去除,点突变质粒-144位和-147位碱基均由A突变为G,但双点突变质粒除了预定的-144位点和-147位点发生突变外,-163位点也发生了突变,由A变为T,序列分析表明,-163不在启动子潜在区域内。
荧光显微镜观察显示(图5),相对于野生型菌株(BL21(DE3)-pET28a-aP-295--101-gfp),所有的定点突变菌株的启动子活性均显著降低。荧光分光光度计测定结果(图6)表明,双点突变(P-295--101(-144、-147Mu))和 2 个 TATA 盒缺失(P-295--101(-116--134dMu))比单点突变(P-295--101(-144 Mu)和P-295--101(-147 Mu))引起的启动子活性降低幅度更大,提示苏云金芽胞杆菌aiiA启动子-150--142 bp区域的2个TATA盒对aiiA的5'端侧翼区域的启动子活性可能起着很关键的作用。
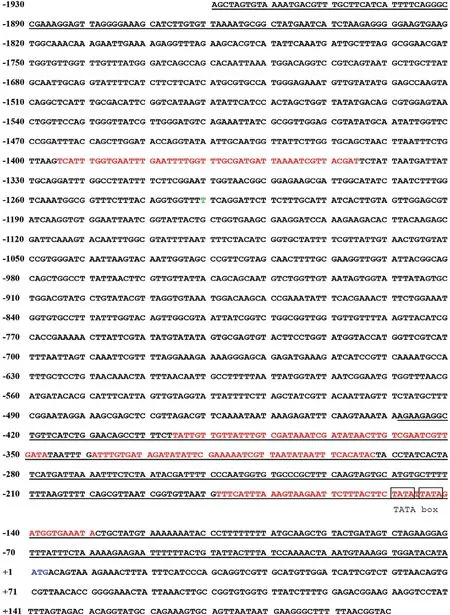
图1 aiiA的5'端侧翼序列
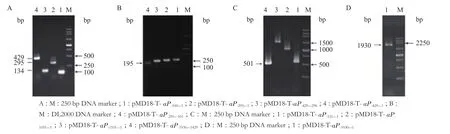
图2 aiiA的5'端侧翼序列的亚克隆
3 讨论
将AiiA蛋白运用于农业生产,提高aiiA的转录水平是至关重要的一步。有研究表明将苏云金芽胞杆菌cry3Aa的启动子用于启动aiiA,可以提高aiiA的表达,增加AiiA蛋白的产量[13]。一些报告基因,如绿色荧光蛋白,常用于研究启动子功能活性的鉴定[14-15]。本实验为研究aiiA的启动子活性,克隆了苏云金芽胞杆菌aiiA的5'侧翼序列(aP-1930--1),通过荧光显微镜和荧光分光光度计对aiiA的5'端侧翼序列的功能进行定性和定量分析,根据报告基因GFP的荧光强度分析表明,分段克隆到的侧翼序列aP-295--101、aP-295--1和aP-1930--1429均具有启动子活性,这与通过生物信息学分析其启动子潜在区域结果一致,但序列aP-1930--1不具备启动子功能。此外,通过在NCBI上BLAST比对发现序列-1 821--1430 bp区域是氨基酸通透性酶家族蛋白(Amino acid permease family protein)基因的编码区,表明aiiA的5'端-1 930--1 429 bp区不是aiiA启动子,在苏云金芽胞杆菌中一个启动子同时启动两个基因表达的可能性不大。分段克隆分析显示侧翼序列aP-295--101的启动子活性比aP-295--1强,而且aP-100--1序列没有启动GFP转录的能力。结果表明,aP-100--1序列可能是负调控序列,这也可能导致AiiA蛋白表达量低。
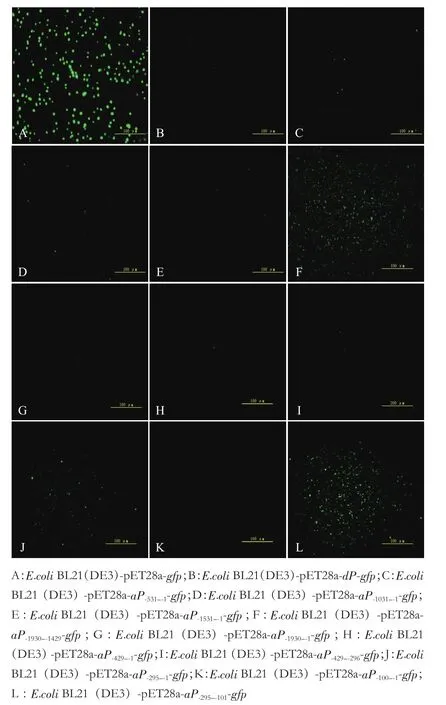
图3 亚克隆片段启动E.coli绿色荧光蛋白GFP的表达
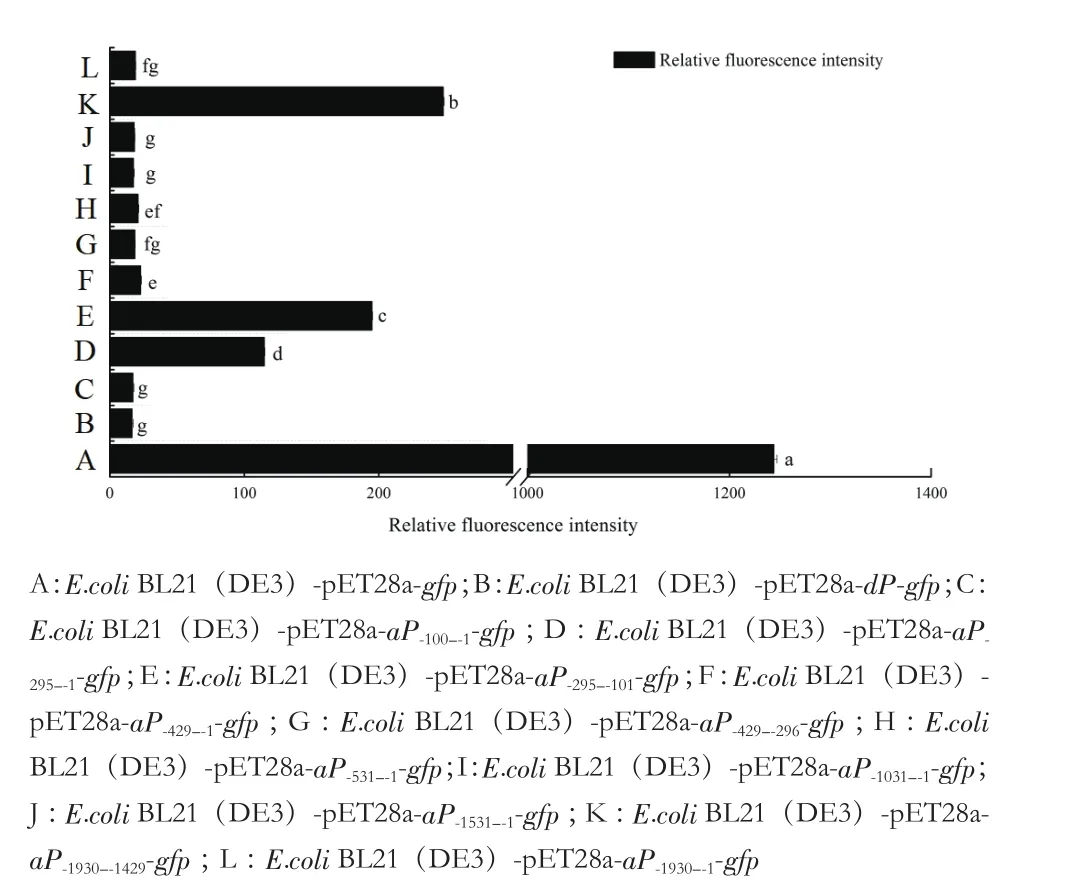
图4 侧翼序列功能鉴定菌株荧光强度值

图5 突变片段启动E.coli绿色荧光蛋白GFP的表达
定点突变是研究基因和启动子功能的有效方法[16-18],前期研究显示,定点突变能够有效的用于蛋白质热稳定性的研究[19]。aiiA定点突变后,表达的AiiA蛋白其酶活力和温度耐受性均得到了提高[20]。本研究以定点突变技术来研究aiiA启动子活性位点,结果表明单点突变使得该基因启动子的活性显著降低,双点突变(P-295--101(-144、-147Mu))和TATA盒缺失进一步降低其活性。
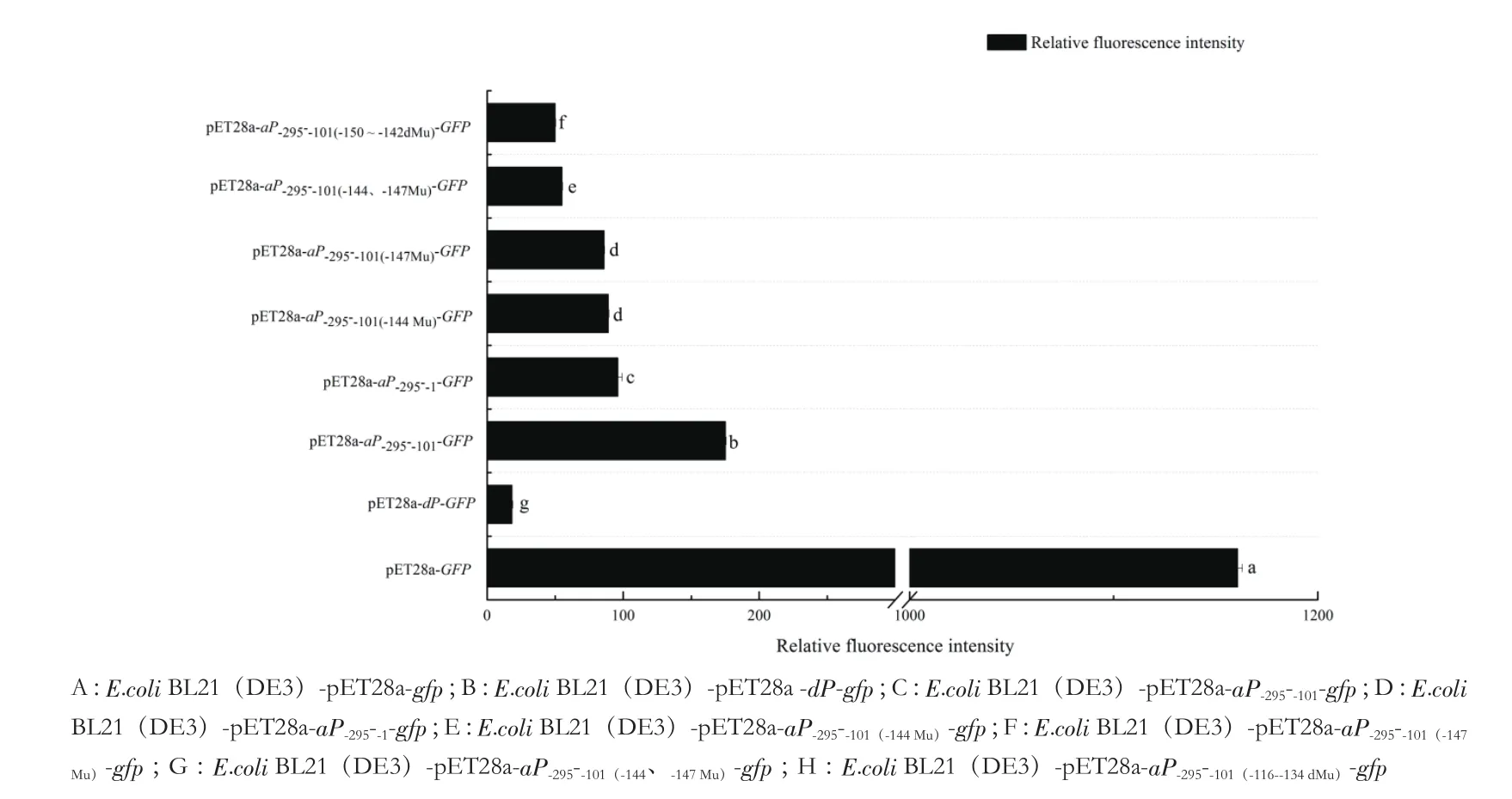
图6 突变菌株荧光强度值
启动子具有启动和调节基因转录的作用[21-123]。传统原核启动子一般位于转录起始位点上游-10区(TATAAT)和-35区(TTGACA),然而本研究发现,位于aiiA转录起始位点上游-100 bp处的序列没有启动子活性(-295--101 bp可能是aiiA的启动子)。有研究表明,苏云金芽胞杆菌的cryIIIA毒素基因的启动子区域至少由基因上游的-635--553 bp,-553--367 bp以及-367-18 bp 3个结构域组成[24]。可能苏云金芽胞杆菌基因的启动子存在距转录起始位点较远的机制。苏云金芽胞杆菌AiiA蛋白表达较低很可能是由aiiA启动子上游和下游序列对aiiA启动子的负调控作用所致。
当然本文使用大肠杆菌替代原始宿主苏云金芽胞杆菌进行研究也存在一些不足,首先不同的表达载体其转录因子可能不同,会对转录调控造成不同的影响;其次,用gfp代替aiiA,可能会由于目的蛋白不同而导致其表达模式的不同,从而影响对启动子功能的判断。尽管如此,本研究根据破囊壶菌Δ4-脂肪酸脱饱和酶基因5'端侧翼区域的研究方法对苏云金芽胞杆菌aiiA的5'端侧翼序列克隆并在大肠杆菌中进行分段表达,其确实可以启动报告基因gfp的表达,具有启动子功能活性,为aiiA基因最可能的启动子区域及其重要的转录元件[25];但其启动aiiA表达的具体调控机制仍需要进行同源性表达分析验证。该结果有利于后续进一步深入研究该启动子在原始宿主中的功能及其转录调控分子机制,也有利于对aiiA在大肠杆菌等异源宿主中进行高表达的研究。
4 结论
苏云金芽胞杆菌aiiA的5'端-295--1 bp为aiiA的启动子区,-429--296与-100--1序列是aiiA的负调控序列。-150--142 bp区域的2个TATA盒对aiiA的5'端侧翼区域的启动子活性起重要作用。
猜你喜欢
环球时报(2022-09-20)2022-09-20 15:18:57
中国生物防治学报(2022年3期)2022-07-09 10:00:22
微生物学杂志(2021年2期)2021-07-01 11:01:06
微生物学杂志(2020年2期)2020-12-31 07:17:13
今日农业(2020年24期)2020-12-15 16:16:00
上海农业科技(2019年3期)2019-06-25 10:45:36
江西农业(2018年9期)2018-08-20 12:34:14
河北林业科技(2016年5期)2016-11-08 03:13:16
兽医导刊(2016年12期)2016-05-17 03:51:50
现代检验医学杂志(2015年4期)2015-02-06 02:02:06
