
皮肤血管肉瘤一例并文献复习
2018-11-29 07:37:04张秋鹂常建民
中国麻风皮肤病杂志 2018年11期
张秋鹂 常建民
皮肤血管肉瘤(cutaneous angiosarcoma,cAS),又称为恶性血管内皮细胞瘤,是一种少见的、起源于内皮细胞的、具有高度侵袭性的恶性肿瘤[1]。该病预后较差,具有较高的复发率和转移率,目前治疗措施有限,因此及时和准确诊断至关重要。现报道1例皮肤血管肉瘤,并对其病因及分子生物学机制、临床特征、病理特征、鉴别诊断及治疗、预后进行文献复习。
1 病例资料
患者,女,66岁。因头皮结节1年,额部瘀斑8个月就诊于我科。患者1年前无明显诱因枕后出现少量丘疹、结节,后皮疹逐渐增多、增大,部分出现破溃、渗液,伴头皮肿胀、疼痛,曾在外院就诊并给予外用抗生素药膏治疗后皮疹无明显缓解,渐发展成浸润性斑块。8个月前右侧额颞部出现水肿性紫红色斑片,皮损面积逐渐增大。4个月前头皮斑块、结节增多,伴局部脱发,头皮疼痛症状加剧。发病期间曾多次就诊,考虑为“头部脓肿性穿掘性毛囊周围炎”,口服头孢类抗生素、盐酸米诺环素,外用夫西地酸等药物治疗,疗效欠佳。既往史:高血压病、2型糖尿病病史10余年,目前口服降压药及降糖药,血压、血糖控制可。否认家族遗传性疾病。皮肤科查体:头皮散在肤色至淡红色结节,表面可见少量鳞屑及毛细血管扩张,伴脱发(图1)。右侧额颞部可见水肿性紫红色斑片,压之不褪色(图2)。皮肤组织病理学及免疫组织化学检查:表皮大致正常,真皮内可见弥漫肿瘤细胞增生,形成不规则腔隙,腔隙内未见明显红细胞,管腔内皮细胞部分有异型性(图3、4)。管腔内皮细胞CD31(+)、CD34(+)、FVIII(+)、Ki67 30%(图5)。PET-CT检查未见淋巴结及远处转移。
诊断:皮肤血管肉瘤。
治疗:明确诊断后,因皮损较大无法行扩大手术切除术,转至放疗科先行放射治疗,仍在随访中。
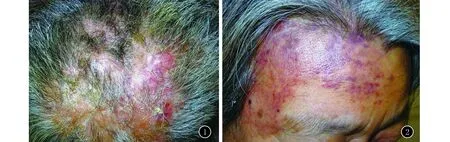
图1 头皮散在肤色至淡红色结节,表面可见少量鳞屑及毛细血管扩张,伴脱发图2 右侧额颞部可见水肿性紫红色斑片,压之不褪色
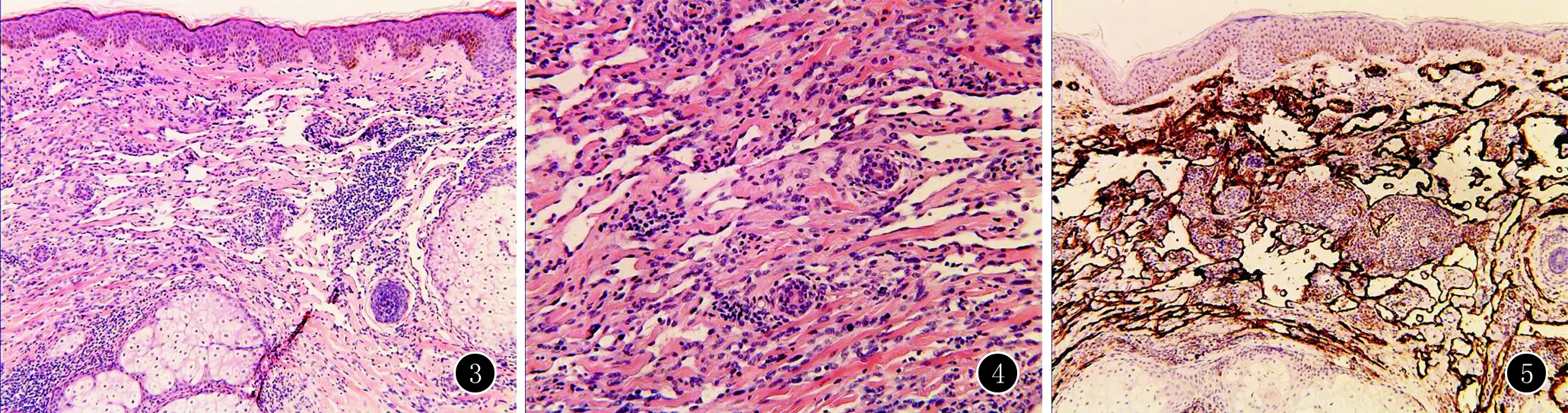
图3 表皮大致正常,真皮内可见弥漫肿瘤细胞增生,形成不规则腔隙(HE,×100) 图4 腔隙内未见明显红细胞,管腔内皮细胞部分有异型性(HE,×200) 图5 管腔内皮细胞CD31(+)(免疫组化染色,×200)
2 讨论
临床分型:皮肤血管肉瘤最常见的类型见于老年人的头颈部,属于原发性皮肤血管肉瘤,又称为Wilson-Jones型,占头颈部恶性肿瘤的0.1%以下[2]。放疗相关性皮肤血管肉瘤是一种少见类型,属于放射治疗的并发症之一,常见于乳腺癌保乳治疗方案的患者,其发病率为0.05%~0.14%。从放射治疗开始至出现皮肤血管肉瘤的潜伏期为2~30年,中位潜伏期为5年[3]。慢性淋巴水肿相关性皮肤血管肉瘤占血管肉瘤的5%左右。大部分报道的病例发生在晚期乳腺癌患者行乳腺切除术后10~15年。先天性淋巴水肿、丝虫病和病理性肥胖的病人亦有继发皮肤血管肉瘤的报道[4-6]。其他的临床类型包括心脏或主动脉血管肉瘤转移至皮肤。儿童皮肤血管肉瘤少见,通常与放疗或遗传性疾病有关,例如着色性干皮病、Aicardi综合征、先天性淋巴水肿。
病因及分子生物学机制:本病发病机制较为复杂,病因包括淋巴潴留、放疗和慢性光接触等。其基因突变位点涉及多个信号传导通路,目前研究发现的突变基因包括TP53、PTPRB、PLCG1[7]。TP53基因突变抑制了肿瘤抑制蛋白p53的合成,有研究发现一半以上的皮肤血管肉瘤的病例存在TP53基因突变[8]。PTPRB和PLCG1是皮肤血管肉瘤的特异性突变,二者具有激活血管生成的作用[9]。近来发现的NUP160-SCL43A3融合基因通过调节其他基因的表达进而影响血管生成[10]。原发性和继发性皮肤血管肉瘤的发生具有不同的分子生物学机制。Manner等[11]发现,在继发性皮肤血管肉瘤中,55%的样本存在8q24.21染色体上MYC基因的高水平扩增,而原发性皮肤血管肉瘤则不存在MYC基因扩增。作为原癌基因,MYC可以调控细胞增殖、分化和凋亡,并可通过上调miR-17-92 cluster促进血管生成。由于皮肤血管肉瘤来源于血管或淋巴管,血管内皮生长因子(VEGF)及其受体(VEGFR)引起了广泛关注。其中,VEGF-A通过其受体VEGFR-1(由FLT1基因编码)及VEGFR-2(又被称为flk或KDR),在血管生成及血管通透性中发挥了重要的调节作用,而VEGF-C和VEGF-D则通过VEGFR-2及VEGFR-3调节淋巴管的生成,并被认为与肿瘤的淋巴转移有关。通过免疫组化分析,发现在血管肉瘤中普遍存在VEGF及其受体的异常激活。
临床特征:本病好发于老年人的头部、面部、颈部,男性更常见,男女发病率比为1.7∶1[1]。皮损表现为良性撞伤样皮损,可伴有面部肿胀和水肿。进一步发展后的皮损表现为紫罗兰色隆起性结节、斑块和肿物。
组织病理学及免疫组织化学特征:皮肤血管肉瘤呈浸润性或侵袭性生长模式,在不同个体之间以及单个瘤体内的内皮细胞间细胞分化程度差异很大,但其病理特征类似。分化好的区域表现为筛状血管网,形成不规则管腔样结构,通常不含血细胞,内皮细胞具有异型性。分化较差的区域管腔不明显,内皮细胞可形成乳头状凸起,细胞异型性更加明显,与其他肉瘤、恶性黑素瘤不易鉴别。不规则管腔样结构、管腔内有红细胞、部分区域有出血现象可作为诊断线索。亦有透明细胞样、印戒细胞样、颗粒细胞样的血管肉瘤病例报道[12]。CD31和CD34是最常用的免疫组织化学染色,大多数血管肉瘤表现为CD31和CD34阳性,CD31具有更强的特异性。血管肉瘤亦对VIII因子、PROX-1、Ulex europaeus等染色阳性。近来研究发现肿瘤细胞高表达CD98预后较差[13]。
本病例皮损性质表现为疼痛性肤色至红色结节,甚至在额部出现皮肤瘀斑之后,仍多次误诊为头部穿掘性毛囊周围炎,因此可能与临床医生对本病不熟悉有关。本病例组织病理学与典型皮肤血管肉瘤相符合,肿瘤细胞形成管腔样结构,细胞具有异型性。由于患者皮损面积较大,与患者家属沟通后,放弃局部切除术,暂行放射治疗,必要时行系统化疗,目前仍在随访中。头面部血管肉瘤的临床表现多种多样,需与外伤后瘀斑、蜂窝织炎、真菌感染、结节病、血管性水肿、血管瘤等相鉴别。本病例早期通常易与头部穿掘性毛囊周围炎鉴别,后者头皮常有反复发生的丘疹、脓疱和脓肿形成。额部出现皮肤瘀斑后需要和系统性皮肤淀粉样变进行鉴别,后者组织病理可见真皮内及血管壁有均一红染、无定形、团块状的淀粉样蛋白沉积,血管周围可见红细胞外溢。本病有时需要与梭形细胞黑素瘤或癌相鉴别,这种情况下主要依靠免疫组化,S-100蛋白是恶性黑素瘤的特异性免疫组化标志,还可以检测HMB-45、Ki-67等。
预后:本病预后较差,其中位生存期为3.4~5年。局部复发率高达63%。远处转移率为36%,肺转移为最常见的远处转移,其次为骨转移、肝转移[14]。皮肤血管肉瘤进展迅速,在所有头颈部的软组织肉瘤中其淋巴结转移率最高。预后差的相关因素包括发病年龄大于50岁、男性、心血管疾病病史、吸烟史、发病部位在头皮、肿瘤直径大于5 cm、诊断时已经出现卫星灶[15]。早期识别、及时诊断对血管肉瘤的治疗很关键。
治疗:目前本病的主要治疗方法为手术联合放射治疗,有研究表明,联合治疗的患者总生存率高于单纯手术治疗或放射治疗[14]。虽然目前没有指南推荐具体的切除范围,但仍推荐扩大切除肿瘤边界3 cm以及深切[3]。化疗对该病的治疗目前仍有争议,有学者认为辅助化疗对该病是有益的,而有的学者认为化疗并不能提高总生存率[14,15]。系统化疗常用于有转移的或者不能行肿瘤完整切除的患者。以阿霉素为基础的化疗方案为治疗软组织肉瘤最常用的化疗方案,可使疾病无进展期达到3个月,含有紫杉醇的化疗方案可以延长疾病无进展期至4~5个月[16,17]。近年来,血管内皮生长因子的抑制剂贝伐单抗已用于该病的治疗,可使疾病无进展期至6.5个月[18]。目前仍在研究的治疗方案包括内因子的单克隆抗体。
猜你喜欢
影像研究与医学应用(2022年23期)2023-01-11 10:52:58
中国卫生产业(2021年17期)2021-09-22 02:39:40
基层中医药(2020年4期)2020-02-13 11:54:49
家庭医学(下半月)(2019年11期)2020-01-16 08:39:08
家庭医学(下半月)(2019年10期)2019-11-16 08:59:52
中国临床医学影像杂志(2019年4期)2019-06-18 10:55:06
实用临床护理学杂志(电子版)(2017年26期)2017-04-01 11:15:17
Coco薇(2015年5期)2016-03-29 23:19:50
中国医疗美容(2015年1期)2015-07-12 10:05:55
郑州大学学报(医学版)(2015年1期)2015-02-27 14:50:36
