
诱导多能干细胞在帕金森病治疗中的应用进展
2018-11-28 09:45陈枕枕牛昱宇
生物技术进展 2018年6期
陈枕枕, 牛昱宇
昆明理工大学生命科学与技术学院, 昆明 650500
帕金森病(PD)是一种神经退行性疾病,患者在晚期阶段伴随静止震颤、身体僵硬、运动障碍等运动性退化症状。其主要病理学特征是中脑黑质(substantial nigra, SN)中多巴胺能神经元(DAns)功能异常。然而,人们对于PD发病机制的认识和有效的治疗手段研究还十分有限,主要原因是缺乏有效的研究模型。
诱导多能干细胞(iPSCs)是类似于胚胎干细胞(embryonic stem cell, ESCs)且具有自我更新和分化潜能的一种干细胞。2006年,Takahashi等[1]在前人研究基础上利用病毒载体将4个转录因子(Oct3/4、Sox2、Klf4和c-Myc)转入体细胞中,使其重编程为一种多能干细胞。次年,Takahashi等[2]又使用类似的方法成功获得了由人的体细胞得到的iPSCs。随着研究的深入,研究人员也得到了非病毒诱导处理得到的iPSCs[3],为iPSCs在基础和临床研究方面奠定了基础。
虽然以小鼠为代表的PD动物模型已得到广泛应用,但仍存在诸多不足:无法体外直接观察;不能重复DAns的变化、路易小体的形成和神经突损失的完整过程;PD患者的尸检仅能观察到最终状态,而不能重现其发病过程。因此,寻找新的能如实反映PD患者发病过程的模型成为研究PD发病机制的关键所在。而来源于患者的iPSCs恰好可以弥补动物模型的不足:方便体外观察;能够完整重现DAns形态功能异常、内部变化的过程;便于控制由环境、动物个体差异等因素带来的干扰,是模拟PD发生的病理固有特征和细胞水平PD研究的最佳选择。此外,基于iPSCs的自体细胞移植治疗也是近年来研究的热门话题。本文将从iPSCs细胞模型、自体细胞移植治疗PD等方面介绍iPSCs在PD研究中的进展,以期为PD的相关研究提供参考。
1 iPSCs与PD细胞模型
研究人员用单基因异常的iPSCs逐步诱导神经祖/干细胞及各类成熟神经元(如DAns、DA前体细胞和星形胶质细胞等),通过控制变量来观察该基因对神经元发育、病变的影响,因此可用iPSCs细胞模型来研究单基因与PD发病的关系,从而了解PD的发病机制。用来研究PD发病机制的iPSCs模型主要有LRRK2突变、PINK1突变、α-synuclein突变、Parkin突变、GBA突变等类型,科学家们正是从这些细胞模型的研究中揭示PD发病的机制。
1.1 LRRK2突变
LRRK2突变主要包括N1437H、R1441C、G2019S 3个位点的突变,其中G2019S是最常见的一种[4]。研究人员在G2019S-iPSC衍生的DAns中发现并报道了氧化应激、SNCA蛋白积聚、线粒体自噬和DNA受损的现象,与正常DAns相比,G2019S-iPSC衍生的DAns对氧化应激和蛋白酶体应激诱导的凋亡更敏感,这些细胞经长期培养后,SNCA蛋白的表达水平明显升高[5,6]。除了氧化应激和SNCA蛋白质累积现象外,在这些DAns中还可以看到神经突数目和分支减少等现象[7],而氧化应激、SNCA蛋白积聚、线粒体自噬以及DNA损伤都是PD的典型病理学特征[8]。
1.2 PINK1突变
PINK1编码线粒体靶向激酶,可以保护神经元免受应激诱导的线粒体功能障碍[9]。2011年,Seibler等[10]通过PINK1-iPSC诱导的DAns发现了线粒体去极化、拷贝数增加和PGC-1表达上调等现象。Rakovic等[11]证实,来自PINK1-iPSC的Parkin蛋白表达水平降低,并且由于泛素化功能障碍导致线粒体膜电位丧失而引起间质性浸润,这表明PINK1可能与线粒体损伤有关。更重要的是,在这些DAns中通过慢病毒转染使其表达PINK1,能够恢复由于Parkin基因缺陷引起的线粒体易位,进一步验证了PINK1和Parkin的协同作用[12,13]。
1.3 α-synuclein突变
α-synuclein是路易小体的主要成分,也存在于SNCA三倍体患者的DAns中[14]。2011年,Devine和Byers报道称,突触核蛋白三倍体(AST)患者和正常人SNCA蛋白的表达量没有差异。但分化成DAns时,与正常神经元相比,AST神经元中SNCA蛋白的数量增加了1倍,并且AST神经元对氧化应激更敏感,进一步证实了SNCA蛋白积累和氧化应激在PD中的重要作用[15,16]。
1.4 Parkin突变
Parkin编码三亚基连接酶复合物的组成部分,其介导底物蛋白质靶向蛋白酶体降解。它除了与PINK1在线粒体损伤和氧化应激中的协同作用外,也与DA的内平衡有关,来自PD患者iPSC衍生的DAns表现出DA吸收降低、释放增加的现象。由于线粒体功能障碍,这些DAns中的活性氧水平也有所提高,这表明Parkin蛋白可以提高DAns神经传递的准确性,抑制DA氧化[17,18],对神经元损伤具有保护作用[19]。
1.5 GBA突变
GBA基因编码溶酶体膜蛋白β-葡萄糖脑苷脂酶(也称为酸性β-葡糖苷酶),其突变会导致糖脂底物在溶酶体中积累。葡萄糖脑苷脂酶抗体和溶酶体功能障碍被认为是PD的重要致病机制[20]。2012年,Panicker等报道,GBA-iPSC衍生DAns显示出SNCA基因高表达,并且由于葡萄糖脑苷脂酶(GCase)缺陷,大鼠噬菌体的清除能力下降[21,22]。
这些细胞模型在体外模拟PD患者的发病过程,直观地反应细胞之间的联系、功能损伤直至完全坏死的过程,但也有其不足之处。目前培养细胞一般是贴壁培养,但细胞在机体内部相互之间联系的紧密程度远远超过培养皿中的情况,所以细胞培养过程中所表现出的联系程度远远不够,还需要更为立体的模型来解决这一问题,如3D培养技术、类脑体的研究等。
2 iPSCs与PD细胞治疗
目前PD的治疗方法主要分为三大类:药物治疗、手术治疗和细胞治疗。药物治疗以左旋多巴类药物为主,这种治疗方法虽然在前期治疗中效果显著,但随着时间的推移,其效果会随耐药性的产生逐渐降低甚至消失,且对患者有很大副作用。手术治疗适用于药物治疗效果不好、病情难以控制的PD患者,但由于风险大、费用昂贵、治疗效果无法保证,因而许多患者无法接受。由于受损细胞种类单一,又分布在特定的局限空间,这些发病特点又为细胞治疗提供了先天条件。
2.1 iPSCs技术与自体细胞移植
自体细胞移植(图1)是采用病人来源于自身的细胞经过体外分化、基因编辑、移植的过程用于疾病治疗的一种手段。目前研究中用于治疗的干细胞主要包括胚胎干细胞、iPSCs、神经干细胞和间充质干细胞,在治疗的过程中不同类型的干细胞展现出了各自的优劣,相比较于其他几类干细胞,iPSCs的来源最为方便、安全,且分化潜能仅次于胚胎干细胞,因此成为细胞治疗的首选。
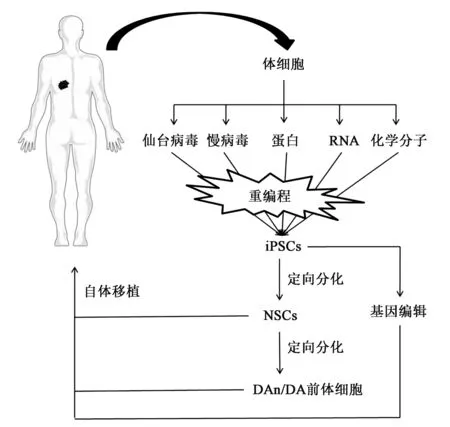
图1 自体细胞移植示意图Fig.1 Transplanting diagram of autologous cells.
Yoshikawa等[23]在大鼠PD模型中的细胞移植实验有力地支持了自体移植的可行性及治疗效果等问题,为一些神经系统功能性损伤及缺失疾病的治疗带来了希望。类似的,中国科学家将猕猴iPSCs诱导出的DAns移植入药物模型猴体内,发现其症状得到了明显改善[24]。2017年,日本科学家在前人的实验基础上,分别使用健康人和PD患者(非基因突变)iPSC诱导的DA前体细胞移植入PD猴模型脑内,并进行了长达两年的观察,发现这些细胞成功地在模型猴体内存活、增殖并发挥正常功能,没有发现致瘤现象,成功缓解了模型猴的症状,并发现PD患者与健康人来源的iPSCs同样具有治疗效果[25]。也就是说,对于非基因突变型患者而言,不需要经过体外修复过程,就可以达到治疗效果。而对于家族性PD患者,则需要通过体外基因编辑对突变基因进行修饰,才可以得到具有正常功能的DAns,达到细胞治疗的效果。
虽然细胞移植治疗PD是比较理想的方法,但也存在诸多不足。首先,在体内分化的DAns是否会因为基因异常而无法正常发挥功能,这需要对基因突变型患者来源的iPSCs进行体外基因修复,然后再诱导分化移植回病人体内,但基因修复过程中会不会引入其他突变,这也是一个悬而未决的问题。其次,人体是一个综合复杂的系统,不同细胞类型之间的联系密不可分,虽然iPSCs可以建立PD细胞模型,但是无法显示其移植后与其他类型细胞相互作用的情况。再次,如何确定移植物的存活、增殖以及是否发挥正常功能。已有研究使用影像学结合同位素示踪的方法来追踪移植物的生存状况[25],但该方法还处于探索阶段,其准确性、时效性还有待提高,距离临床应用还有很长一段路要走。
2.2 细胞自体移植存在的问题
2.2.1细胞来源 最初的iPSCs是通过病毒导入4个多能性基因来实现的,其中包括致癌基因c-Myc,但致癌基因的重新启动对于临床治疗来说是一个潜在的风险,科学家们为此也付出了大量努力试图避开致癌基因的使用。2009年,Lin等[26]通过SB43125 和PD0325901两个化学小分子得到了人iPSCs,这解决了人们担心的细胞来源安全性的问题。之后,小分子重编程体系也不断完善,更多的小分子被用于重编程中,如Forskolin、CHIR99021、DZNep、RepSox[27]等抑制信号通路型小分子,以及Staerk[28]发现的SOX2替代物RepSox 616452、LY-364947、Dasatinib、iPYrazine、PP1等。2015年,Li等[29]用ISX9、I-BET151、CHIR99021、Forskolin 4个小分子将小鼠的成纤维细胞转分化得到神经元。同年,Hu等[30]也通过ValproicAcid、CHIR99021、RepSox、Forskolin化学小分子将阿尔兹海默患者的成纤维细胞转分化为神经元。
虽然目前还没有报道研究化学方法诱导的DAns,但在未来细胞移植中,化学来源的细胞(如DAns、DA前体细胞、NSCs等)致癌风险小,且小分子容易被细胞代谢,是目前已知多种细胞来源里面最为安全的,但DAns的诱导还需更多的研究来实现。
2.2.2细胞功能 对于家族性PD患者来说,其发病主要是基因突变引起的,因此,通过体外基因编辑技术纠正患者突变基因的表达,得到正常的DAns/DA前体细胞也是细胞治疗的关键。
近年来,以锌指核酸酶类转录激活因子效应物(transcription activator-like effector nuclease, TALEN)、锌指核酸酶(zinc-finger nuclease, ZFN)以及规律重复短回文序列簇(clustered regulatoryinterspaced short palindromic repeat, CRISPR)为主的基因编辑技术不仅成功应用于斑马鱼[31,32]、啮齿类[33,34]以及非人灵长类[35,36]等模式动物中,还广泛应用于iPSCs的修饰,用于得到基因突变的细胞模型[37,38]、修饰异常基因的表达[39~41]等方面。这些研究的顺利进行使得体外修饰患者异常基因表达成为可能,也为临床应用打下基础。
综上所述,得到非癌变DAns/DA前体细胞,并通过基因编辑技术修饰其突变基因,才可以使iPSCs的应用真正离临床治疗更进一步。
3 展望
虽然目前的研究发现已经取得了很多成果,但是由于PD的发病机制非常复杂并且尚不完全清楚,所以在PD的治疗中,仅依靠iPSCs是非常局限的,必须要与其他生物技术相结合,如基因编辑技术、细胞移植技术等。细胞移植技术的迅猛发展也为PD的治疗提供了解决之道,并且在某些疾病研究领域也获得了重大发现与发展[42]。但是PD的病理进程错综复杂,给临床应用带来了很大阻力。一方面,iPSCs或由其衍生而来的神经细胞移植到患者体内后的存活、生长、迁移及是否发挥功能仍有待研究;另一方面,移植到病患体内的细胞的安全性更需通过各类动物模型来进行更深层次的研究。
猜你喜欢
今日农业(2022年13期)2022-09-15
中华实用诊断与治疗杂志(2022年1期)2022-08-31
海洋通报(2021年1期)2021-07-23
生物学通报(2021年4期)2021-03-16
今日农业(2020年24期)2020-12-15
科学(2020年4期)2020-11-26
生物学通报(2020年10期)2020-08-13
知识经济·中国直销(2017年10期)2017-11-07
中西医结合心脑血管病杂志(2016年20期)2016-03-01
上海农业学报(2016年5期)2016-02-10
