
水环境下氢氧根水分子簇催化缬氨酸旋光异构及羟自由基致其损伤机理
2018-11-28 12:18:52闫红彦王佐成杨晓翠
吉林大学学报(理学版) 2018年6期
张 新, 佟 华, 闫红彦, 王佐成, 杨晓翠
(1. 白城师范学院 物理与电子信息学院, 吉林 白城 137000; 2. 白城师范学院 计算机科学学院, 吉林 白城 137000)
缬氨酸(Val)是生命体内必需的氨基酸, 根据构象和旋光性的不同, 分为左缬氨酸(S-Val)和右缬氨酸(R-Val).S-Val具有活性, 能促进机体发育、 协调神经系统, 可用于动物饲料添加剂[1].R-Val用于合成氯氟戊菊酯[2]、 生化研究及抑制纤维细胞生长[3], 作为手性药物原料, 可用于合成抗肿瘤药物[4].
Val分子旋光异构目前已引起人们广泛关注. 文献[2]研究表明, 醛催化下S-Val可在羧酸中旋光异构; 文献[5-6]研究表明, 高浓度的质子易使Val分子消旋; 文献[7-8]研究表明, Val分子旋光异构裸反应有4个通道,α-H以氨基N为桥迁移是优势通道, 水分子簇对质子转移反应有较好的催化作用; 文献[9]研究表明, 激发态的α-H以羰基氧或氨基氮为桥迁移, 实现了S-Val旋光异构; 文献[10]研究表明, 以氨基氮为质子迁移桥梁的旋光异构具有优势, 水溶剂环境下水分子簇的催化, 使优势通道的决速步骤能垒降为113.24 kJ/mol. 在优势通道中, 羟自由基水分子簇致缬氨酸损伤的能垒为25.34 kJ/mol, 水溶剂效应使损伤能垒提升至83.81 kJ/mol, 表明生命体内缬氨酸可缓慢地实现旋光异构, 羟自由基的存在可致其损伤.
水是重要的溶剂, 大气中富含水蒸气且存在氢氧根, 生命体也是富水环境并存在羟自由基和氢氧根[11], 当氨基与羧基间为分子内单氢键和双氢键时, 氨基酸分子构象稳定[12]. 基于此, 本文研究水环境下(气相和液相), 具有氨基和羧基间单氢键和双氢键的Val在氢氧根水分子簇作用下的旋光异构, 以及羟自由基在新的通道致Val损伤机理.
1 计算方法
在B3LYP[13]/6-31+G(d,p)水平优化水环境下氢氧根水分子簇催化缬氨酸旋光异构及羟自由基致其损伤反应的水气相驻点结构, 计算溶剂化构象时, 将水视为连续介质, 采用自洽反应场(SCRF)理论的SMD模型方法[14]. 通过对过渡态[15]进行内禀反应坐标(IRC)[16]计算, 验证其为连接所期望的局域极小点. 采用微扰理论的MP2方法[17], 在MP2/6-311++G(2df,pd)水平, 计算高水平反应过程体系的单点能. 利用
Gtotal=ESP+Gtc
计算总自由能, 并绘制反应过程的势能面, 其中ESP和Gtc分别为单点能和Gibbs自由能热校正值. 水溶剂环境下,S型Val分子的构型1与其前面/后面的氢氧根水分子簇通过氢键作用形成的氢键配合物, 记作S-Val_1·[(OH-)·H2O(m/n)]@water(m和n分别表示氢氧根水分子簇在Val分子的前面/后面与其配合), 其他体系表示法类似. 计算均由Gaussian09[18]程序完成.
2 结果与讨论
具有氨基和羧基间分子内单氢键及双氢键的Val分子手性对映异构体的几何构象如图1所示. 计算结果表明,S-Val_1和S-Val_2的单点能分别为-401.605 78,-401.605 51 a.u.,S-Val_1比S-Val_2略稳定. 研究表明,S-Val的旋光异构可在通道a和b实现: 通道a为氢氧根水分子簇与α-H和氨基通过氢键作用形成底物, 氢氧根抽取α-H; 通道b为氢氧根水分子簇与α-H和羰基O通过氢键作用形成底物, 氢氧根抽取α-H; 在通道a和b中均可实现羟自由基致S-Val损伤, 水溶剂对其影响较小.
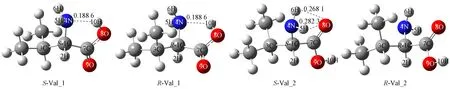
图1 S型和R型缬氨酸分子的几何构象Fig.1 Geometric conformation of S-type and R-type of valine molecules
2.1 氢氧根水分子簇催化Val_1旋光异构
2.1.1 通道a内的旋光异构 氢氧根水分子簇催化Val_1在通道a旋光异构的反应过程如图2~图4所示, 反应过程的Gibbs自由能势能面如图5所示. 对于非氢迁移过程第一、 二、 五、 六基元, 水溶剂对驻点构象影响较小, 因此仅给出气相的构象, 反应机理见文献[10], 本文仅讨论第三和第四基元反应.
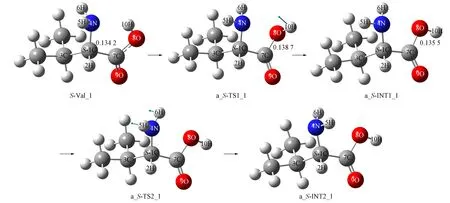
图2 氢氧根水分子簇催化S-Val_1在通道a旋光异构的第一、 二基元反应Fig.2 Elementary reactions of the 1st, 2nd of optical isomerism of hydroxyl water clusters catalyzed S-Val_1 in channel a
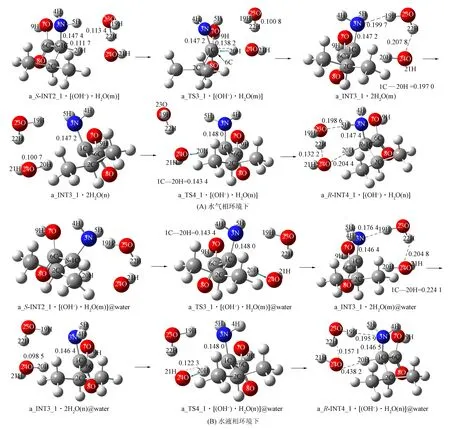
图3 氢氧根水分子簇催化S-Val_1在通道a旋光异构的第三、 四基元反应Fig.3 Elementary reactions of the 3rd, 4th of optical isomerism of hydroxyl water clusters catalyzed S-Val_1 in channel a
1) 经第一基元反应,S-Val_1异构成a_S-INT1_1, 羧基从反式平面结构变为顺式平面结构, a_S-INT1_1与正面的氢氧根水分子簇形成氢键无空间位阻.
2) a_S-INT1_1经第二基元反应, 异构成略稳定的中间体a_S-INT2_1.
3) 第三基元. 氢氧根水分子簇在20H和5H的前面与S-INT2_1通过氢键作用形成配合物a_S-INT2_1·[(OH-)·H2O(m)], 经过渡态配合物a_TS3_1·[(OH-)·H2O(m)], 实现20H从α-C向氢氧根24O—21H的迁移, 形成产物配合物a_INT3_1·2H2O(m). 该基元反应氢氧根抽取α-H形成水分子, 水分子19H—23O—22H未参与反应, 仅具有稳定氢氧根空间位置的作用.
a_TS3_1·[(OH-)·H2O(m)]七元环结构的氢键键角1C…20H…24O,24O…22H—23O和23O…5H—3N分别为169.89°,173.04°,155.88°, 接近平角, 水分子和氢氧根形成较强氢键, 因此a_TS3_1·[(OH-)·H2O(m)]较稳定. 该过程中23O—22H从0.113 4 nm缩小至0.100 8 nm, 释放少许能量; 反应活性中心骨架二面角3N-1C-2C-6C由128.17°变为136.67°, 形变微小; 1C—3N从0.147 4 nm缩小至0.147 2 nm, 释放少许能量. 1C—20H从0.111 7 nm增加至0.138 2 nm, 与文献[10]结果相比, 其拉伸幅度减小, 且仅有一个化学键拉伸, 所需能量大幅度减小. 因此, a_TS3_1·[(OH-)·H2O(m)]产生的能垒较低, 仅为11.45 kJ/mol.
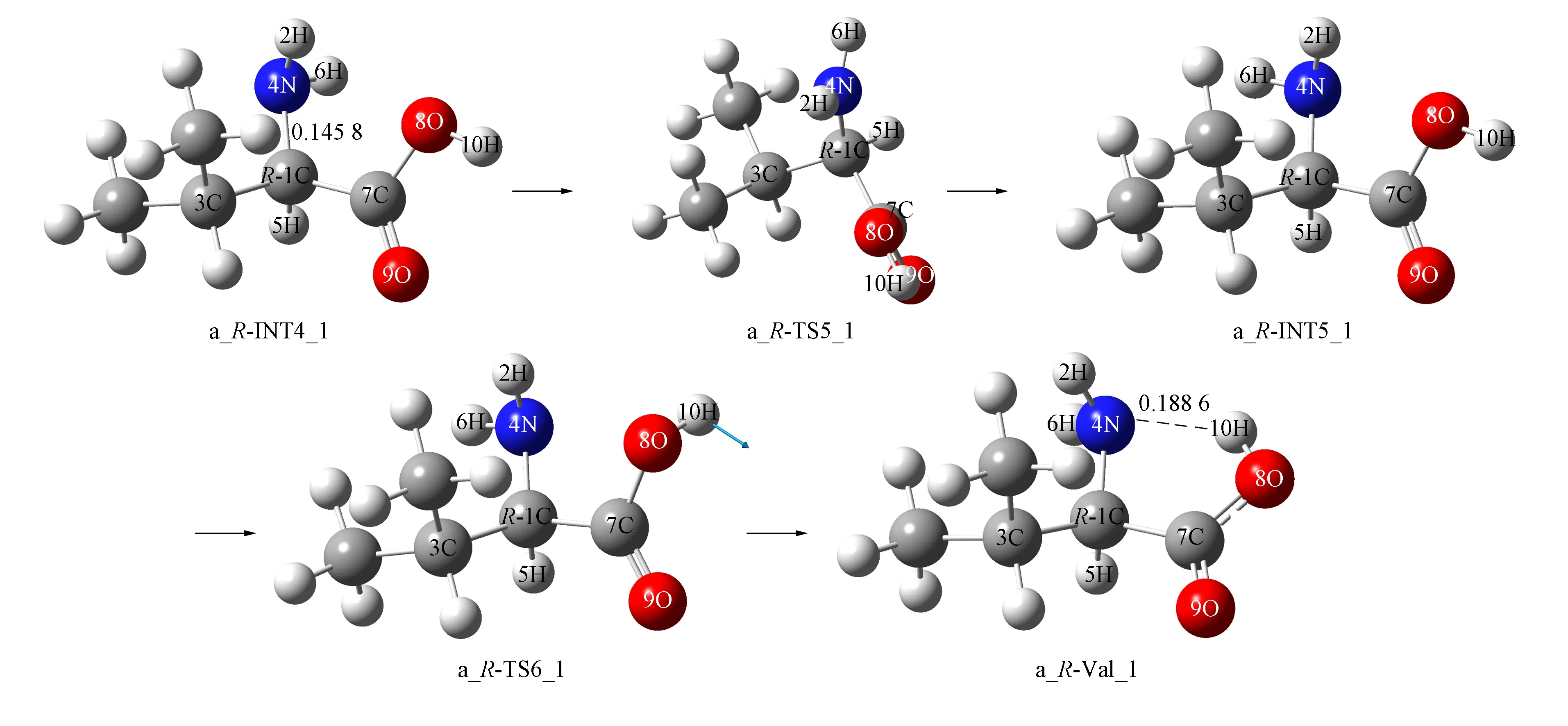
图4 氢氧根水分子簇催化S-Val_1在通道a旋光异构的第五、 六基元反应Fig.4 Elementary reactions of the 5th, 6th of optical isomerism of hydroxyl water clusters catalyzed S-Val_1 in channel a
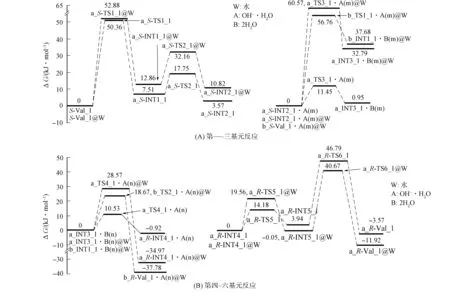
图5 氢氧根水分子簇催化Val_1旋光异构反应的Gibbs自由能势能面Fig.5 Gibbs free energy potential energy surface of optical isomerism reaction of Val_1 catalyzed by hydroxyl water clusters
结构分析表明, a_INT3_1·2H2O(m)的水分子间以及水分子和氨基间为强氢键, 水分子和α-C也形成氢键, a_INT3_1的3N-1C-6C-7O-8O形成离域的大π键, 因此a_INT3_1·2H2O(m)较稳定. 但由于α-C为sp2杂化, 非满配, 因此在势能面上a_INT3_1·2H2O(m)处于比a_S-INT2_1·[(OH-)·H2O(m)]略高的位置. a_INT3_1的α-C失去手性, 导致缬氨酸损伤, 即经过该基元反应后S-Val_1损伤.
水溶剂化效应使该基元反应能垒升高至60.57 kJ/mol. 能垒大幅度提升的原因为: ① 在水液相环境下, 反应活性中心骨架二面角3N-1C-2C-6C由129.65°变为139.07°, 1C—3N从0.146 4 nm拉伸至0.148 0 nm, 骨架形变增大; ② 1C…20H从0.109 6 nm增至0.143 4 nm, 拉伸幅度增加; ③ a_S-INT2_1·[(OH-)·H2O(m)]的偶极矩(6.182 6)大于a_TS3_1·[(OH-)·H2O(m)]的偶极矩(3.647 6), 在极性溶剂水中, a_S-INT2_1·[(OH-)·H2O(m)]比a_TS3_1·[(OH-)·H2O(m)]体系能量降低的程度大. 由于a_INT3_1·2H2O(m)的偶极矩(2.513 3)小于a_S-INT2_1·[(OH-)·H2O(m)]的偶极矩(6.182 6), 因此在极性溶剂水中, a_INT3_1·2H2O(m)比a_S-INT2_1·[(OH-)·H2O(m)]能量降低的程度小.
4) 第四基元为第三基元的逆反应, 2个水分子簇在a_INT3_1的后面与3N和1C通过氢键作用形成配合物a_INT3_1·2H2O(n), 经过渡态a_TS4_1·[(OH-)·H2O(n)], 实现20H从水分子20H…24O…21H向α-C的迁移, 形成产物配合物a_R-INT4_1·[(OH-)·H2O(n)]. 在该基元反应中, 水分子19H—23O—22H未参与反应, 起到稳定另一个水分子20H—24O—21H空间位置的作用, 为α-C抽取20H—24O—21H上的质子20H创造了条件. 从a_INT3_1·2H2O(n)到a_TS4_1·[(OH-)·H2O(n)]过程, 24O—20H从0.100 7 nm增加至0.125 7 nm, 拉伸幅度为0.025 0 nm, 小于第三基元的反应物至过渡态1C…20H的增幅0.026 5 nm, 因此, a_TS4_1·[(OH-)·H2O(n)]小于a_TS3_1·[(OH-)·H2O(n)]产生的能垒, 仅为10.53 kJ/mol.
结构分析表明, a_R-INT4_1·[(OH-)·H2O(n)]具有两个分子间强氢键和一个分子间弱氢键, 且其α-C为sp3杂化, 处于满配状态, 使a_R-INT4_1·[(OH-)·H2O(n)]比a_INT3_1·2H2O(n)处于势能面略低的位置. 水溶剂化效应使该基元反应的能垒升高至28.57 kJ/mol, a_R-INT4_1·[(OH-)·H2O(n)]@water相对于a_INT3_1·2H2O(n)@water的能量大幅度降低, 原因与第三基元反应类似.
经第三、 四基元反应, 完成了α-H从纸面外向纸面内的净迁移,S-INT2_1实现了旋光异构, 即S-Val_1实现了旋光异构.
5) a_R-INT4_1经第五、 六基元反应异构成更稳定的产物a_R-Val_1, 结构分析表明, a_R-Val_1为S-Val_1的手性对映体.
2.1.2 通道b内的旋光异构 氢氧根水分子簇催化Val_1在通道b的旋光异构反应过程如图6所示, 反应过程的Gibbs自由能势能面如图5所示. 本文仅讨论水液相环境下的反应机理.

图6 氢氧根水分子簇催化S-Val_1在通道b的旋光异构Fig.6 Optical isomerism of hydroxyl water clusters catalyzed S-Val_1 in channel b
1) 第一基元. 氢氧根水分子簇在S-Val_1的前面与20H和8O通过氢键作用形成前驱配合物b_S-Val_1·[(OH-)·H2O(m)]@water, 前驱配合物经过渡态配合物b_TS1_1·[(OH-)·H2O(m)]@water, 形成中间体产物配合物b_INT1_1·2H2O(m)@water, 氢氧根抽取α-H形成水分子. 该基元反应与通道a的第三基元反应相似, 水分子19H—23O—22H未参与反应, 仅起到稳定氢氧根空间位置的作用. 结构分析表明: 过渡态b_TS1_1·[(OH-)·H2O(m)]@water的八元环结构基本共面, 3条氢键均较强, 其结构稳定; 从b_S-Val_1·[(OH-)·H2O(m)]@water到b_TS1_1·[(OH-)·H2O(m)]@water过程, 反应活性中心骨架二面角3N-1C-2C-6C由123.88°变为135.38°, 1C—3N键长从0.147 6 nm缩小至0.147 4 nm, 骨架形变较小, b_TS1_1·[(OH-)·H2O(m)]@water产生的能垒较低. 但1C…20H从0.109 6 nm拉伸至0.143 6 nm, 需要一定的能量, 因此b_TS1_1·[(OH-)·H2O(m)]@water产生了56.76 kJ/mol的能垒. 结构分析表明, b_INT1_1·2H2O(m)@water具有水分子间及水分子与b_INT1_1间的强氢键, 并具有水分子与b_INT1_1间的弱氢键, 因此b_INT1_1·2H2O(m)@water为较稳定的中间体配合物.
2) 第二基元. 2个水分子簇在b_INT1_1的后面与α-C和羰基O通过氢键作用, 形成中间体反应配合物b_INT1_1·2H2O(n)@water, 经过渡态配合物b_TS2_1·[(OH-)·H2O(n)]@water, b_INT1_1·2H2O(n)@water的b_INT1_1抽取水分子20H—24O—21H上的质子20H, 形成产物配合物b_R-Val_1·[(OH-)·H2O(n)]@water. 从b_INT1_1·2H2O(n)@water到b_TS2_1·[(OH-)·H2O(m)]@water过程, 1C—3N键长从0.145 9 nm拉伸至0.147 4 nm, 24O…20H键长从0.098 9 nm拉伸至0.121 2 nm, 幅度较小. 反应活性中心骨架二面角3N-1C-2C-6C由163.19°变为135.38°,α-C从sp2杂化(空配状态)向sp3杂化(满配状态)过渡, 是放热过程. 因此, b_TS2_1·[(OH-)·H2O(n)]@water低于b_TS1_1·[(OH-)·H2O(m)]@water产生的能垒, 仅为18.67 kJ/mol.
经第一、 二基元反应,α-H在氢氧根水分子簇与2个水分子簇的作用下, 实现了从纸面外向纸面内的净迁移,S-Val_1在通道b实现了旋光异构, 即S-Val_1在通道b实现了手性对映体转变.
由图5可见, 在水液相环境下, 氢氧根水分子簇催化Val_1旋光异构反应的优势通道为通道b, 决速步骤能垒为56.76 kJ/mol, 通道a的决速步骤能垒为60.57 kJ/mol. 这两个能垒均远小于水分子簇催化Val_1在这两个通道中旋光异构的决速步骤能垒110.61,156.43 kJ/mol, 表明氢氧根水分子簇对Val_1旋光异构反应的催化作用明显优于水分子簇, 尤其在通道b中氢氧根水分子簇显示了极好的催化作用. 56.76,60.57 kJ/mol均小于质子迁移温和反应能垒84.00 kJ/mol[19], 因此, 当生命体内存在大量氢氧根时, Val_1会较快地旋光异构, 严重影响健康. 水气相环境下,S-Val_1在通道a旋光异构的决速步骤为第一基元反应, 能垒为50.36 kJ/mol, 由于该通道的正反应和逆反应能垒均较低, 因此通道a中反应物、 损伤产物、 旋光异构产物及其他中间体可平衡共存.
2.2 氢氧根水分子簇催化Val_2旋光异构
1) 通道a. 氢氧根水分子簇催化Val_2在通道a旋光异构反应过程如图7(A)所示, 反应势能面如图8所示. 其机理与Val_1在通道a旋光异构过程的第三、 四基元反应相似, 即在羟自由基水分子簇与2个水分子簇的作用下,α-H实现从纸面外向纸面内的净迁移, 故略.
由图8可见, 决速步骤为第一基元反应, 水气相环境的决速步能垒为9.24 kJ/mol, 水液相环境下该能垒升高至65.24 kJ/mol, 表明水溶剂对该反应的阻碍作用较大. 水气相环境下第一、 二基元逆反应能垒分别为5.17,9.24 kJ/mol, 容易越过, 反应物、 中间体和产物共存. INT1_2的α-C失去手性, INT1_2已损伤的Val_2.
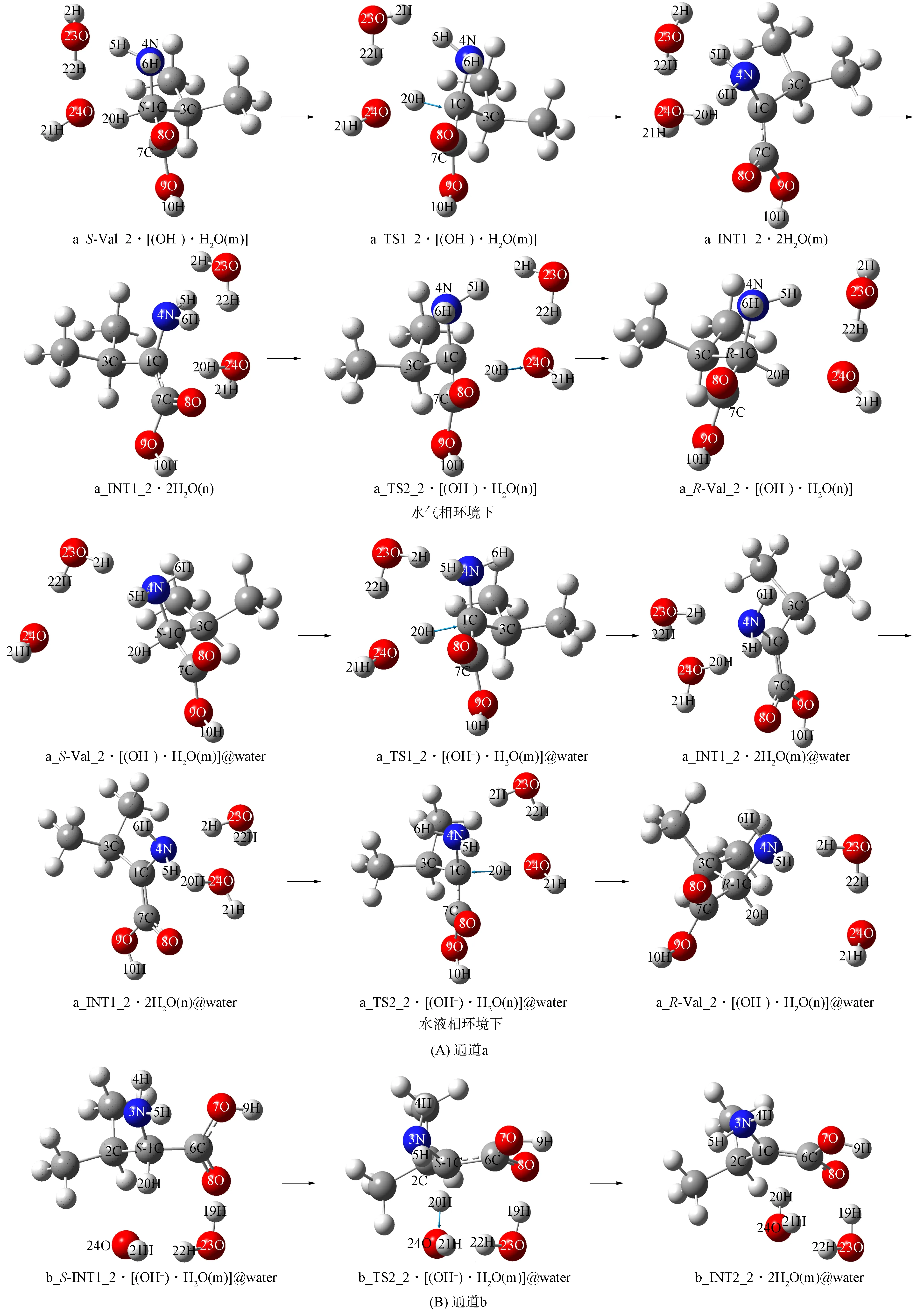
图7 氢氧根水分子簇催化S-Val_2的旋光异构过程Fig.7 Optical isomerism of hydroxyl water clusters catalyzed S-Val_2
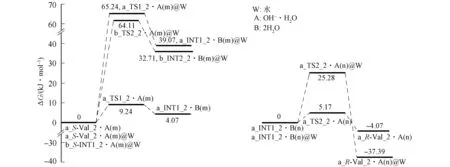
图8 通道a氢氧根水分子簇催化Val_2旋光异构反应的Gibbs自由能势能面Fig.8 Gibbs free energy potential energy surface of optical isomerism reaction of Val_2 catalyzed by hydroxyl water clusters in channel a
水气相环境的决速步骤能垒为9.24 kJ/mol, 远低于质子迁移温和反应能垒84.00 kJ/mol, 表明水汽环境下, Val_2会迅速损伤和消旋. 水液相环境下的决速步骤能垒65.24 kJ/mol也低于质子迁移温和反应的能垒84.00 kJ/mol[19], 表明Val_2在生命体内水环境下也会较快地消旋.
2) 通道b. 水液相环境下氢氧根水分子簇催化Val_2在通道b的旋光异构反应过程如图7(B)所示, 反应势能面如图8所示. 首先, 第一基元反应, 水分子簇作为氢迁移媒介, 10H从9O迁移至8O, 形成中间体b_S-INT1_2, 反应能垒为34.97 kJ/mol[11]. 其次, 第二基元反应(相似于Val_1在通道b旋光异构的第一基元), 氢氧根水分子簇与b_S-INT1_2的α-H及新羰基氧9O通过氢键作用形成的配合物b_S-INT1_2·[(OH-)·H2O(m)]@water, 经过渡态b_TS2_2·[(OH-)·H2O(m)]@water, 氢氧根抽取α-H, 形成b_INT2_2·2H2O(m)@water, 该基元反应为决速步骤, 能垒为64.11 kJ/mol. 最后, 第三基元反应为α-C在另一侧抽取水分子的质子, Val_2在通道b完成旋光异构, 相似于Val_1在通道b旋光异构的第二基元反应, 故略.
2.3 在通道b中羟自由基水分子簇致Val损伤
1) Val_1. 羟自由基水分子簇致Val_1在通道b损伤的反应过程如图9(A)所示, 反应势能面如图10所示.
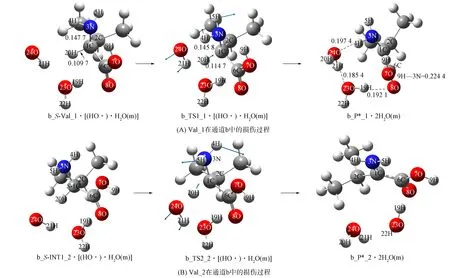
图9 羟自由基水分子簇致Val在通道b的损伤过程Fig.9 Damage process of Val induced by water clusters of hydroxy radicals in channel b
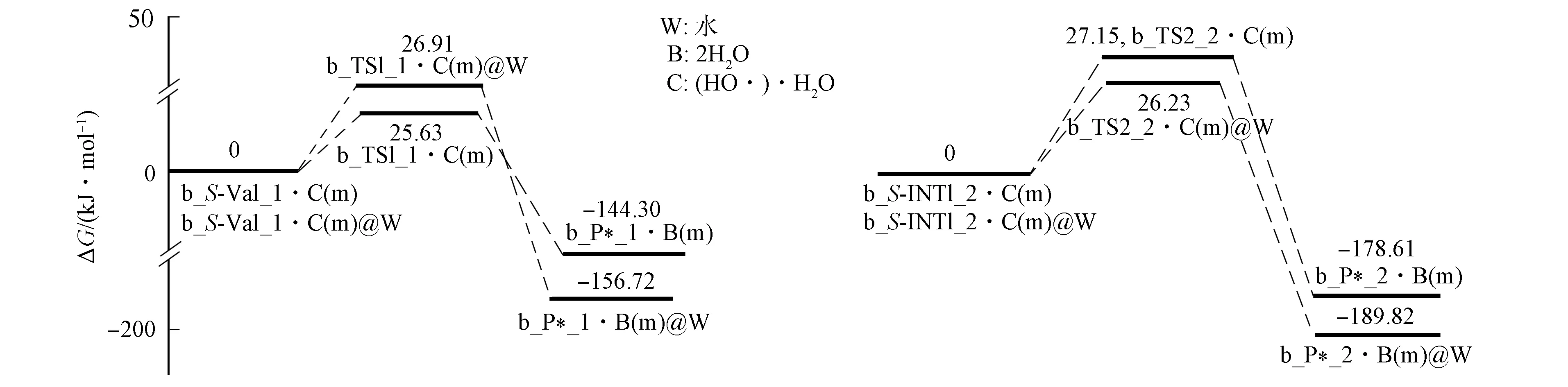
图10 羟自由基水分子簇致Val在通道b损伤反应的Gibbs自由能势能面Fig.10 Gibbs free energy potential energy surface of Val damage induced by water clusters of hydroxy radicals in channel b
首先, 羟自由基水分子簇在S-Val_1的前面, 通过与20H和8O的氢键作用形成前驱配合物b_S-Val_1·[(HO·)·H2O(m)]. 其次, 经过渡态配合物b_TS1_1·[(HO·)·H2O(m)], 羟自由基抽取Val_1的α-H形成水分子, 得至产物配合物b_P*_1·2H2O(m)(*表示产物为损伤的氨基酸, 下同). b_P*_1的α-C失去手性, 即P*_1是已损伤的Val_1产物. 该基元反应与通道a的第三基元反应相似, 水分子19H—23O—22H未参与反应, 仅起到稳定羟自由基空间位置的作用. 结构分析表明: 过渡态b_TS1_1·[(HO·)·H2O(m)]的八元环结构基本共面, 3条氢键均较强, 其结构稳定; 从b_S-Val_1·[(HO·)·H2O(m)]到b_TS1_1·[(HO·)·H2O(m)]过程, 反应活性中心骨架二面角3N-1C-2C-6C由123.62°变为124.43°, 骨架基本未形变, 1C—3N键长从0.147 7 nm缩小至0.145 8 nm, 释放能量, 1C…20H键长从0.109 7 nm拉伸至0.114 7 nm, 幅度较小, 所需能量低. 因此, b_TS1_1·[(HO·)·H2O(m)]产生的能垒较低, 仅为25.63 kJ/mol. 结构分析表明, b_P*_1·2H2O(m)具有3条较强的分子间强氢键及1条分子内单氢键, 骨架原子3N,1C,6C,7O,8O形成五中心离域大π键. 因此, b_P*_1·2H2O(m)结构稳定.
水溶剂化计算表明, 水溶剂效应使损伤能垒升至26.91 kJ/mol, 但损伤产物配合物变得更稳定了, 水溶剂效应对羟自由基的水分子簇致Val_1在通道b的损伤仅有微弱的阻碍作用, 可忽略.
2) Val_2. Val_2在通道b损伤的反应过程如图9(B)所示, 反应势能面如图10所示. 羟自由基水分子簇与b_S-INT1_2的α-H及新羰基9O通过氢键作用形成配合物b_S-INT1_2·[(HO·)·H2O(m)], 经过渡态b_TS2_2·[(HO·)·H2O(m)], 羟自由基抽取α-H, 形成b_P*_2·2H2O(m). 机理与Val_1在通道b的损伤相似, 故略. 计算表明, 水气相环境下的损伤能垒为27.15 kJ/mol, 水溶剂效应使该能垒降至26.23 kJ/mol, 水溶剂仅起微弱的助催化作用.
3 结 论
综上, 本文可得如下结论:
1) 缬氨酸的旋光异构可在2个通道a和b实现, 通道a为氢氧根水分子簇与α-H和氨基通过氢键作用形成底物, 氢氧根抽取α-H后,α-C在另一侧抽取水分子的H; 通道b为氢氧根水分子簇与α-H和羰基通过氢键作用形成底物, 氢氧根抽取α-H后,α-C在另一侧抽取水分子的H. 在通道b中, 水分子辅助羟自由基抽取α-H可致缬氨酸损伤.
2) 水液相环境下, 构象Val_1旋光异构的主反应通道为通道b, 决速步骤能垒为56.76 kJ/mol; 构象Val_2在两个通道a和b旋光异构的能垒分别为65.24,64.11 kJ/mol. 表明生命体内的氢氧根会导致缬氨酸较快地旋光异构, 且Val_1的旋光异构具有优势.
3) 水气相环境下, Val_1和Val_2在通道b的损伤能垒分别为25.63,27.15 kJ/mol, 水溶剂效应对损伤能垒影响较小, 表明生命体内的羟自由基会导致缬氨酸较快地损伤.
猜你喜欢
燃料化学学报(2023年3期)2023-03-11 03:34:40
云南化工(2021年8期)2021-12-21 06:37:38
中学课程辅导·教学研究(2021年8期)2021-07-14 13:44:52
燃料化学学报(2021年5期)2021-06-02 14:01:38
质量技术监督研究(2018年1期)2018-03-26 08:04:28
猪业科学(2017年2期)2017-03-30 09:33:29
光学精密工程(2016年2期)2016-11-07 09:02:32
猪业科学(2016年12期)2016-04-24 01:21:02
饲料博览(2016年3期)2016-04-05 16:07:52
饲料工业(2016年22期)2016-01-10 02:56:06
