
Effects of the ultrasound-assisted H3PO4 impregnation of sawdust on the properties of activated carbons produced from it
2018-11-01 05:21ZHANGZongboLIUXiaoyangLIDaweiGAOTiantianLEIYuqiWUBaoguiZHAOJiaweiWANGYankuiWEILing
新型炭材料 2018年5期
ZHANG Zong-bo, LIU Xiao-yang, LI Da-wei, GAO Tian-tian ,LEI Yu-qi, WU Bao-gui, ZHAO Jia-wei, WANG Yan-kui, WEI Ling
(1. College of Mechanical and Electrical Engineering, China University of Petroleum (East China), Qingdao 266580, China;2. State Key Laboratory of Heavy Oil Processing, China University of Petroleum (East China), Qingdao 266580, China)
Abstract: The effects of the ultrasound-assisted H3PO4 impregnation of sawdust on the pore structure, morphology, surface functional groups, and adsorption capacities for iodine and methylene blue of activated carbons produced from it were investigated. Results showed that ultrasonic impregnation promoted the mass transfer and uptake of H3PO4, which simultaneously increased specific surface area, pore volumes of micropores, mesopores and ultramicropores, and adsorption capacities for iodine and methylene blue. Long ultrasonic impregnation times were unnecessary. The maximum surface area (1 504 m2/g) was achieved at a total impregnation time of 45 min with 5 min ultrasound irradiation. Compared with impregnation without the ultrasound treatment, the ultrasonic-assisted impregnation reduced the impregnation time by 85%.
Key words: Activated carbon; Ultrasound; Impregnation; H3PO4 activation
1 Introduction
Activated carbons (ACs) have been extensively used in purification[1-3], catalysis[4,5], capacitors[6-8]and gas storage[9-11], owing to their highly developed porosity, large surface area, and abundant surface functional groups. The commonly-used methods for preparation of ACs include physical activation and chemical activation. The chemical activation with phosphoric acid, which has been applied industrially, usually involves impregnation before pyrolysis. The impregnation condition together with feedstock type and activation condition can be changed to tune the properties of the H3PO4-activated carbons. The impregnation is often conducted by leaving the H3PO4-feedstock mixtures alone or stirring them, which makes the impregnation time-consuming (usually cost 2 to 24 h)[3,12,13].
Ultrasound can enhance mass transfer and produce mechanical and chemical effects in liquid-solid system[12,14,15], because of acoustic cavitation[16,17]which means formation, growth, and implosive collapse of micro-bubbles in liquid. Ultrasound has extensively been employed to assist adsorption[18-21]and regeneration[22-25]of ACs. However, the use of ultrasound for AC preparation remains scarce. For example, a few researchers prepared ACs by ultrasonic-assisted impregnation of biomasses with alkali solution[26,27]. Nevertheless, the effects of introduction of ultrasound to acid impregnation on properties of ACs are unknown. Given that acid activators are quite different from alkali ones in nature, it is necessary to investigate the effects.
This study aims to investigate the effects of ultrasonic-assisted H3PO4impregnation on properties of resulting ACs. The ACs were prepared from sawdust by H3PO4impregnation under ultrasound irradiation for different times. The effects were discussed in terms of pore structure, yield, surface morphology, surface functional groups, iodine number, and methylene blue value. We found that introduction of ultrasound to H3PO4impregnation increased porosity and adsorption performance of ACs simultaneously, and significantly reduced total the impregnation time required to achieve the maximum iodine number.
2 Experimental
2.1 Materials
Sawdust was dried at 110 ℃ to a constant weight. Its C content, H content, and particle size were 47.1 wt%, 6.4 wt%, and 0.22-0.90 mm, respectively. All chemicals used (H3PO4, HCl, I2, etc.) were analytical reagents.
2.2 Preparation of ACs
To prepare ultrasonic-impregnated AC, 3.5 g of sawdust was first soaked in 28 mL of 15 wt% H3PO4solution under ultrasound (20 kHz, 540 W) for different times (0, 2.5, 5, 10 and 15 min) using an ultrasonic processor (FS-600N, China), then the sawdust-H3PO4mixture was allowed to stand until this standing period plus the ultrasonic-assisted soaking time was 45 min. Afterwards, the impregnated sawdust was dried in an oven at 110 ℃ for 2.5 h. The dried mixture was heated in a tube furnace at 10 ℃ /min from 200 to 425 ℃ and held for 120 min under flowing N2of 400 mL/min. The as-obtained carbonized sample was cooled to room temperature and named C-x-y, where x denoted the ultrasonic-assisted soaking time in minutes and y denoted the total impregnation time. Subsequently, the carbonized sample (C-x-y) was washed repeatedly with distilled water until the elute pH reached about 7.0, and finally dried at 110 ℃ for 10 h. The resulting AC was denoted as AC-x-y. For example, AC-5-45 min was the AC prepared using 45 min impregnation in total, which consisted of 5 min ultrasonic impregnation and 40 min stationary impregnation. For another, the AC-5-5 min was prepared using 5 min ultrasonic impregnation, without conducting stationary impregnation.
2.3 Characterization
The yield of ACs was calculated from yield =m/m0×100%, wherem0andmare the mass of dried sawdust and resulting AC, respectively. The washing loss for ACs was determined by washing loss = (mc-m)/mc×100%, wheremcis the mass of the carbonized samples before washing. This parameter could reflect the amount of P-containing species removed from the carbonized samples by washing.
The31P nuclear magnetic resonance (NMR) spectra of H3PO4solution before and after ultrasonic treatment were analyzed using a NMR Spectrometer (Bruker AVANCE III 600, Germany). The chemical shift was internally referenced to 85 wt% H3PO4.
Nitrogen adsorption/desorption isotherms were obtained using a static volumetric analyzer (Micromeritics, ASAP 2460, USA). Prior to the measurements, sawdust was vacuum-dried at 100 ℃ for 12 h, and ACs were vacuum-dried at 250 ℃ for 5 h. The specific surface area (SBET) was calculated by the Brunauer-Emmet-Teller (BET) equation. The relative pressure range used for calculation of theSBETwas from 0.05 to 0.2. The correlation coefficient and C value were higher than 0.999 9 and 0, respectively. The micropore volume (Vmic) was measured by Dubinin-Astakhov (DA) method. The total pore volume (Vtot) was estimated from the amount of N2adsorbed at relative pressure (p/p0) of 0.99. The pore size distribution was determined by applying nonlocal density functional theory (NLDFT) to adsorption isotherm. Iodine number and methylene blue value of ACs were determined according to China standards GB/T 12496.8-2015 and GB/T 12496.10-1999, respectively. The AC mass and iodine concentration used to determine the iodine number were 0.500 0 g and 0.097 74 mol/L, respectively. Surface morphology of ACs and chemical compositions (C, O, and P) of carbonized samples were analyzed by a scanning electron microscopy (ZEISS MERLIN Compact, Germany) equipped with an energy dispersive X-ray spectrometer (EDAX, XM2-60S). The FTIR spectra of ACs in wavenumber range of 4 000 to 400 cm-1with a resolution of 4 cm-1were recorded on a FTIR spectrometer (Bruker Tensor 27, Germany) by the KBr pellet method.
3 Results and discussion
3.1 Effects on surface area and pore volume
The N2adsorption/desorption isotherms of the obtained ACs are presented in Fig. 1a. The adsorption isotherms sharply increased at low relative pressures (p/p0<0.1), levelled off at relative pressures of 0.3-1.0, and showed no obvious hysteresis loops. These features indicated that the isotherms belonged to the type Ib (IUPAC classification). Thus, the obtained ACs were all microporous, which was also reflected by the abundant pores below 0.7 nm and in the range of 1 to 2 nm (Fig. 1b). However, the pore structure of the ACs was different, as shown by the observation that the ACs displayed different N2adsorption isotherms and pore size distribution curves. Hence, the introduction of ultrasound during H3PO4impregnation affected the pore parameters of ACs, which was further discussed below.
As shown in Table 1, the ACs prepared by ultrasonic impregnation (AC-2.5-45 min to AC-15-45 min) showed higherSBET,Vtot,Vmic,Vmes, andVultrathan AC-0-45 min, indicating that ultrasonic impregnation increased surface area and pore volume simultaneously. To check this promotion effects, we compared pore parameters for AC-5-45 min and AC-5-5 min (Table 1). TheSBETandVtotof AC-5-5 min accounted for 85% and 83% of the corresponding values of AC-5-45 min. This result also showed that the surface area enlargement and pore development were mainly due to ultrasound-assisted impregnation rather than stationary impregnation. Another interesting observation was that as the ultrasonic time increased from 0 to 15 min, theSBETandVtotfor ACs prepared by ultrasonic impregnation (AC-0-45 min to AC-15-45 min) generally firstly increased and then decreased. More specifically, increasing the ultrasonic time from 0 to 5 min enhanced theSBETandVtotby 28.88% and 34.48%, respectively, whereas prolonging the time from 5 to 15 min reduced the SBETandVtotby 7.31% and 11.54%, respectively. This variation trend ofSBETandVtotgenerally well agreed with the change of iodine number with the rise in ultrasonic time (to be presented in section 3.4).

Fig. 1 (a) N2 adsorption/desorption isotherms and (b) DFT pore size distributions of ACs.

SamplesSBET (m2/g)Pore volume (cm3/g)VtotVmicVmesVultraAC-0-45 min11670.580.430.150.079AC-2.5-45 min14560.750.540.210.104AC-5-45 min15040.780.550.240.101AC-10-45 min13210.670.480.190.093AC-15-45 min13940.690.480.220.094AC-5-5 min12730.650.470.180.086
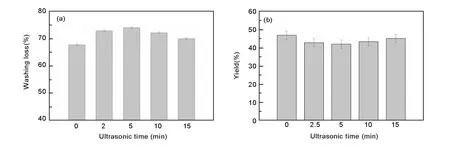
Fig. 2 (a) Washing loss and (b) yield for activated carbons.
During activation, H3PO4can catalyze bond cleavage reactions for the biopolymers in sawdust, crosslink/connect the biopolymer fragments via cyclization and condensation, and form phosphate and polyphosphate bridges[28-31]. Consequently, the acid occupies spaces, hinders material shrinkage, and yields a porous structure after removal by washing with water[32]. Given the above activation mechanism, the aforementioned effects of ultrasonic impregnation on pore parameters were considered relevant to the amount of P-containing species in the carbonized samples (C-x-y series)[33, 34]. To check these explanation, we obtained washing loss and phosphorus content of the carbonized samples, as shown in Fig. 2a. The samples prepared by ultrasonic impregnation showed higher washing loss than the sample fabricated without ultrasonic impregnation, indicating that more P-containing species were incorporated into the precursor in the case of ultrasonic impregnation. This indication was also supported by the observation that carbonized samples having undergone ultrasonic impregnation (C-5-45 min and C-15-45 min, Table 2) showed higher P content than the sample prepared without ultrasonic impregnation (C-0-45 min). When a larger quantity of P-containing species were removed from the carbonized samples by washing, more pores were created, which increased the surface area and pore volume of ACs. Nevertheless, when the ultrasonic period increased from 5 to 15 min, the P-containing species incorporated in the carbonized samples became fewer and fewer, as shown by the decreased washing loss (Fig. 2a). Thus, the number of created micropores declined, which reduced surface area and pore volume of ACs. Additionally, ACs prepared with and without ultrasonic impregnation displayed similar yield (Fig. 2b), suggesting that release of volatile matters during preparation was not the main reason for pore development[33].

Table 2 Surface chemical composition of carbonized samples.

Fig. 3 31P NMR spectra of H3PO4 solution before and after ultrasonic irradiation for different times.
To discuss the effects of ultrasonic impregnation on the quantity of P-containing species incorporated in the precursor, we conducted NMR analysis for H3PO4solution and pore parameter analysis for the sawdust. As shown in Fig. 3, the NMR spectra for H3PO4solution before and after ultrasonic irradiation were similar in that they all showed a peak at chemical shift of (0.688-0.700)×10-6, which was ascribed to the phosphorus in H3PO4molecule. This result indicated that ultrasonic irradiation did not change the chemical form of H3PO4in the solution. Thus, the effects of ultrasonic impregnation on the quantity of P-containing species were not due to the ultrasonic-induced change in chemical form of H3PO4. Besides, the effects of ultrasonic impregnation were not attributable to the ultrasonic-induced variation in pore parameter of the sawdust, because theSBETof the sawdust did not change markedly after the sawdust was irradiated with ultrasound for 5 min in distilled water (1.5 vs. 0.4 m2/g).
At this moment, it is necessary to consider cavitation effects. The propagation of ultrasound in sawdust-H3PO4mixture can produce transient cavitation bubbles and steady ones. The collapse of transient cavitation bubbles resulted in jet pulses and acoustic steaming. These effects enhanced mass transfer near solid surface[35,36]via causing inter-particle collision, structural breaking, and formation of micro-cracks on solid surface[26,27]. As a result, more H3PO4molecules could infiltrate and/or react with the sawdust, which increased the quantity of P-containing species in the carbonized samples. Conversely, when the ultrasonic time was increased above 5 min, more steady cavitation bubbles might be attached to the sawdust surface, which hindered the contact of sawdust with H3PO4. Hence, the quantity of P-containing species in the carbonized samples decreased.
To check the explanation presented above, two carbon samples named C-0-45 min-noP and C-5-45 min-noP were prepared respectively by the same way that AC-0-45 min and AC-5-45 min were fabricated, except that no H3PO4was added in water during the impregnation. The sample C-5-45 min-noP was similar to C-0-45 min-noP inSBETandVtot(245 vs. 268 m2/g, and 0.11 vs. 0.12 cm3/g), which indicated that ultrasonic impregnation did not increaseSBETandVtotof carbons when no H3PO4was used during impregnation. This result agreed with our statement that the effects of ultrasonic impregnation on the quantity of P-containing species in the carbonized samples were related to the variation in H3PO4mass transfer near the sample surface.
It should be mentioned that the highestSBETfor the ACs prepared by ultrasonic impregnation was 1 504 m2/g (Table 1). Such a value was higher compared with the surface areas reported for biomass-based H3PO4-activated carbons (Table 3). This observation indicated that ultrasonic impregnation was a promising technique for preparing H3PO4-activated carbons with large surface areas.

Table 3 Comparison with reported biomass-based H3PO4-activated carbons.
3.2 Effects on morphology
SEM images of AC-0-45 min and AC-5-45 min are shown in Fig. 4. The two samples displayed tube-like micrometer-sized pores and resembled each other in surface morphology. This resemblance suggested that ultrasonic impregnation hardly affected morphology of H3PO4-activated carbons at the micron scale.

Fig. 4 SEM images of (a) AC-0-45 min and (b) AC-5-45 min.
3.3 Effects on surface functional groups


Fig. 5 FTIR spectra for ACs prepared by ultrasonic impregnation for different times.
3.4 Effects on iodine number
Iodine number can reflect the ability of ACs to adsorb small molecules from liquid[40]. As shown in Fig. 6a, iodine number of ACs prepared using ultrasonic impregnation was higher than that of AC-0-45 min, indicating that ultrasonic impregnation enhanced the ability of ACs to adsorb small molecules like iodine from wastewater. This enhancement was understandable, as iodine adsorption depends on micropore volume and surface area[40], which were both increased by ultrasonic impregnation (Table 1).
Besides, the total impregnation time for the sample with the highest iodine number (AC-5-45 min, 1 113 mg/g) was 45 min, whereas the period required for achieving similar iodine number was as long as 300 min when no ultrasound was introduced during impregnation (Fig. 6). These results indicated that introduction of ultrasound during impregnation reduced total impregnation time by 85%, which was highly favorable for efficient production of H3PO4-activated carbons. The reduction in impregnation time was also reflected by the fact that AC-5-5 min showed a higher iodine number (1 018 mg/g > 923 mg/g) than AC-0-45 min but was prepared within a much shorter total impregnation time (5 min < 45 min). The reduction in total impregnation time was attributable to the ability of ultrasonic-assisted H3PO4impregnation to increaseSBETandVmicof ACs.

Fig. 6 Iodine number of activated carbons prepared by impregnation in (a) presence and (b) absence of ultrasound.
3.5 Effects on methylene blue value
Methylene blue value is often used to evaluate ability of ACs to remove organic compounds like dye from wastewater. As shown in Fig. 7, ACs prepared by ultrasonic impregnation all showed higher methylene blue value than AC-0-45 min, indicating that ultrasonic impregnation increased the ability of AC to remove dye molecules from wastewater. Methylene blue molecule, whose minimum molecular cross-section is about 0.8 nm, can enter pores with pore diameter larger than 1.3 nm[41]. Thus, its adsorption mainly occurs in large-micropores and mesopores. The ACs fabricated by ultrasonic impregnation possessed more large-micropores and mesopores than AC-0-45 min, as shown by the pore size distribution curves (Fig. 1b) andVmes(Table 1). These characteristics made the ACs form ultrasonic impregnation display superior adsorption performance for methylene blue. Among the ACs made by ultrasonic impregnation, AC-5-45 min showed the highest methylene blue value, because it had the largest number of large micropores and mesopores, as suggested by its pore size distribution in pore range of 1.3 to 4 nm (Fig. 1b).

Fig. 7 Methylene blue value for ACs prepared by ultrasonic impregnation for different times.
4 Conclusion
Introduction of ultrasound during H3PO4impregnation simultaneously improvedSBET,Vtot,Vmic,Vmes,Vultra, iodine number, and methylene blue value of ACs, but long-time ultrasonic irradiation was unnecessary. The parameters generally firstly increased and then decreased with increasing ultrasonic-assisted impregnation time, and reached their respective maxima when the ultrasonic-assisted impregnation lasted for 5 min. The obtained maximumSBETwas 1 504 m2/g, which ranked among the high specific surface areas ever reported for biomass-based H3PO4-activated carbons. The introduction of ultrasound during H3PO4impregnation considerably shortened the total impregnation time required for achieving the maximum iodine number. The FTIR analysis indicated that ultrasonic-assisted impregnation promoted the formation of O-containing groups in ACs.

- 新型炭材料的其它文章
- 碳基功能材料在海洋领域中的应用进展
- A two-step method for the preparation of high performance corncob-based activated carbons as supercapacitor electrodes using ammonium chloride as a pore forming additive
- 稻壳基活性炭负载镍催化剂的制备及在香草醛加氢脱氧反应中的催化性能
- Preparation and process optimization of randomly oriented C/C composites by a novel method
- Analysis of the interaction energies between and within graphite particles during mechanical exfoliation
- 负载辛伐他汀的氧化石墨烯/丝素蛋白屏障膜的制备及其生物学性能