数字PCR在功能核酸精准检测中的研究进展
2018-10-26 02:12:24刘晓朱鹏宇王垚朱水芳付伟
生物技术通报 2018年9期
刘晓 朱鹏宇 王垚 朱水芳 付伟
(中国检验检疫科学研究院,北京 100176)
随着分子生物学的快速发展,人们发现并合成了一类具有一定的特殊功能的核酸分子,包含但不限于转基因产品中的外源插入片段、微生物中的致病基因、核酸适配体(Aptamer)及脱氧核酶(DNAzyme)等,人们把这一类对生物体或体外检测反应具有特殊功能的核酸分子叫做功能核酸[1-3]。功能核酸的出现开拓了人们的视野也加深了理解,普遍意识到核酸除了作为遗传信息的载体和运转载体以外还可以广泛应用:转化事件插入产生新型性状表达,微生物入侵寄主后致病基因的增殖与表达,核酸适配体与脱氧核酶的出现为非核酸物质检测的发展提供广阔空间[4-6],同时还有医疗诊断[7-8]等领域。功能核酸的开发对人类认知疾病类型以及开发新型诊断技术有着重大价值与意义。
PCR是一种体外酶促扩增特异性DNA的实验技术。从1985年美国科学家 Kary Mullis开发了第一代以PCR技术为基础进行定性分析的实验方法后[9],PCR方法被不断改进:由定性的分析方法发展到定量测定,从原先只能扩增几个kb的基因到已能扩增几十个kb的DNA片段。到目前为止,PCR技术已有十几种之多。在这种背景下,第三代基于分割体系的绝对定量PCR应运而生,其中通过有限稀释法和根据泊松分布统计原理来准确计算DNA浓度的方法,称为数字PCR(Digital PCR,dPCR)。1992年dPCR初具雏形,Sykes等[10]使用有限稀释、PCR和泊松分布的方法在非淋巴细胞和正常体细胞的背景下检测到了低丰度lgH重链突变基因,进行了精细的定量研究,但当时并没有明确指出dPCR这一个概念。直到1999年Vogelstein等[9]首次建立了dPCR基本实验流程和重要原则:以终点信号的有无作为判断依据,正式提出了dPCR的概念。Vogelstein等通过对样品的有限稀释,检测了结肠癌患者粪便样品中KRAS基因突变的同时提出了dPCR的一种优势:稀释提高了对反应抑制剂的耐受程度。2003年,Devin等[11]又提出了BEAMing技术(珠子、乳剂、扩增和磁力),利用包被链霉亲和素的磁性珠子和生物素标记的寡核苷酸形成微乳液进行PCR扩增,该技术为今后的微滴式dPCR的产生奠定一定的基础。本文将对数字PCR技术及其在现阶段不同领域中的功能核酸检测研究现状进行归纳和综述,并对数字PCR未来的发展前景进行展望,旨为促进数字PCR在功能核酸检测领域进一步的应用。
1 数字PCR
数字PCR是一种基于单分子扩增以及原始反应分割的一种PCR扩增手段。通过将原始的反应体系进行有限的大量分割,将参与扩增的组分和体系中的模板分配到单个的反应小体系中。通过PCR的扩增,当小的反应体系中存在目标分子时,会产生扩增产物并释放出荧光信号,反之,当体系中不存在目标分子时,该小反应体系为阴性。后续进行数据分析时,需要根据所有体系中的荧光值确定阳性体系与阴性体系的数量,从而根据两者数量的比较,计算出原始体系中所含有的目标基因的绝对拷贝数[12-13](图 1)。
当原始体系中模板的含量相当少时,理论上每一个阳性体系内部只含有单个目标分析,此时,最终结果的阳性亮点数就可以近似等于体系中的目标分子数。但是大部分情况下,样品中目标分子的量不会太低,此时单个阳性体系中的目标分子的数量可能会达到2个,甚至更多。这时就需要通过泊松分布(Poisson distribution)的原理来计算体系中的总的目标分子的拷贝数。根据泊松分布的原理,反应体系中目标分子的拷贝数可以通过公式1计算[14]:
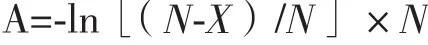
其中,A为反应体系中目标分子的拷贝数,X为数字PCR的阳性体系数,N为总体系数。
通过公式可以看出,随着反应阳性体系数(X)的增加,体系中目标分子的拷贝数相对于X会有较大的差距,随着X的持续增加,数字PCR结果的不确定度也随着提高,总体来说,数字PCR阳性体系的数量不得超过总体系数量的80%。另一个方面,N的增加会使整个数字PCR体系具有较大的线性范围,并可以提高反应的灵敏度、稳定性以及可重复性。
在常规的定量PCR实验中,引物的扩增效率会对反应产物量以及定量结果产生较大的影响[15]。但是对于数字PCR来说,最终实验结果的分析只取决于最终所有小体系的荧光信号而与引物的扩增效率几乎没有关系。所以对于数字PCR来说,实验结果很少会受到引物设计因素的影响。
综上所述,相对于常规的定量PCR,数字PCR具有极强的绝对定量能力,可以不依赖于标准曲线和标准物质直接对样品进行目标分子的定量,并且具有很高的灵敏度、稳定性以及可重复性。可以很好的应用在各个领域的核酸分子绝对定量检测中。
2 商业化数字PCR平台及其特点
发展到现在,市面上常见的dPCR主要有两种:微滴式dPCR(droplet dPCR,ddPCR)和芯片式dPCR(chip dPCR,cdPCR)技术(图1)。
cdPCR主要以Fluidigm公司的BioMark HD系统以及Life Technology公司的QuantStudio 3D系统为代表(图2)。Bio Mark系统[16]采用微泵阀式芯片,以聚二甲基硅氧烷为芯片材料,主要依靠微流控通道与阀门的开闭进行原始体系分割,在芯片的反应仓进行PCR反应,然后通过类似于基因芯片的方法扫描每个通孔的荧光信号,进行目的序列含量的计算。QuantStudio 3D系统[17]采用阵列微池式芯片,反应液由进样孔直接进入各微反应池。芯片式dPCR生成微滴体积均一,具有较高的稳定性,体系之间影响较小,但技术操作复杂,通量有限且实验成本较高。
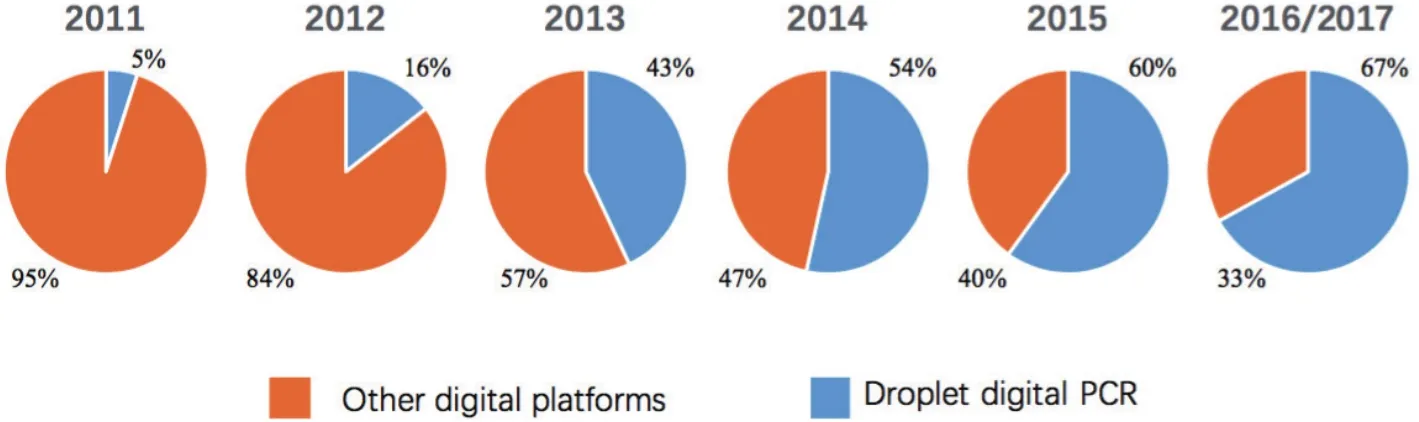
图1 2011年-2017年数字PCR平台应用变化趋势
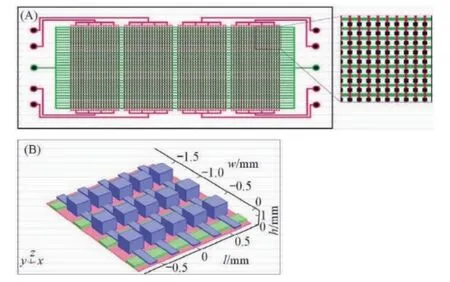
图2 芯片式数字PCR平台宏观结构(A)微观结构(B)
ddPCR主要有Bio-rad公司开发的QX200系统和Rain Drop系统,QX200可以将体系分割成2万个微滴,Rain Drop可以分割成100万-1 000万个微滴[18](图3)。ddPCR原理是通过将一个待分析的PCR反应体系进行微滴化处理,利用微滴发生器制成近20 000个油包水小微滴从而对原始体系进行分割,样品中的核酸分子随机分配到大量独立的微滴中,每个微滴中含有一个或不含待检核酸分子。对微滴体系进行扩增反应以后,分析每个微滴的荧光信号,进行有或无的判断,将判断结果按照泊松分布的原理,通过读取靶标和内参核酸的阳性微滴个数以及比例从而得到靶分子的拷贝数及浓度。与芯片式dPCR相比,操作简单,可以实现高通量的检测,也保证一定程度上的微滴检测稳定性。
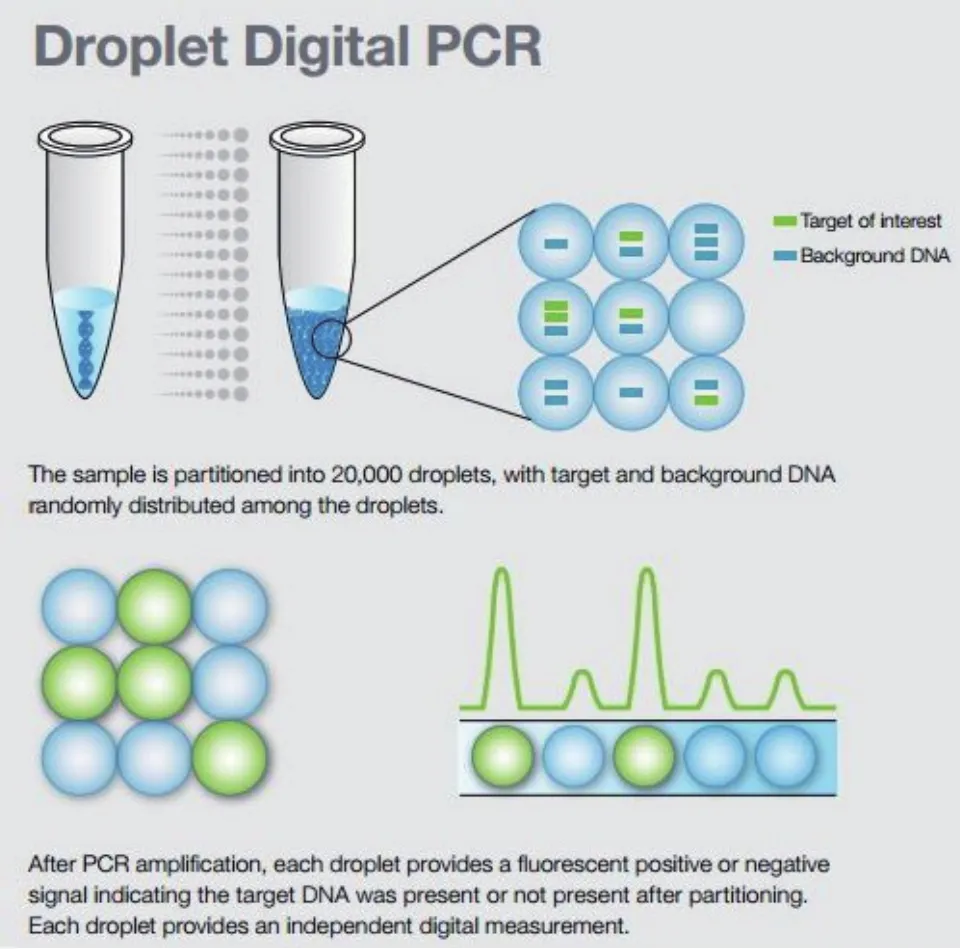
图3 微滴式数字PCR原理
与常规的PCR的方法相比,ddPCR有很好的优势。(1)绝对定量。常规PCR和实时荧光PCR定量检测都需要已知拷贝数的标准DNA制定标准曲线,由于样品测定在各种条件上不会完全一致,会造成PCR扩增效率的差异,从而影响定量结果的准确性。而ddPCR不受标准曲线和扩增动力学影响,可以进行绝对定量。(2)样品需求量低。在检测珍贵样品和样品核酸存在降解时具有明显的优势。(3)高灵敏度。ddPCR本质上是将一个传统的PCR反应分成了数万个独立的PCR反应,在这些反应中可以精确地检测到很小的目的片段的差异、单拷贝甚至是低浓度混杂样品,且能避免非同源异质双链的形成[19]。(4)高耐受性。由于目的序列被分配到多个微滴中,显著降低了体系间的影响以及背景序列和抑制物对反应的干扰,扩增基质效应大大减小。尽管dPCR技术具备较好前景,但由于96孔板或384孔板加样的复杂操作为精确测量带来了困难,且相较于实时荧光PCR来说成本较高,距广泛应用还有一定距离。
3 数字PCR在转基因外源插入片段检测中的应用
3.1 转基因产品成分检测与鉴定
商业作物育种一般通过常规方法或遗传转化引入所需性状,尤其是不存在于植物基因组的新特性。目前随着市场转基因产品的逐步释放,我国必须有涵盖转基因的插入、稳定性及性状表达等所有方面的详尽档案。目前被大部分国际组织认可的标准转基因定量检测方法是基于Taqman探针的实时荧光PCR,但dPCR作为新兴、准确的技术受到越来越多人的青睐。基于Taqman探针的ddPCR是在同一个孔内同时进行内参基因与目的基因的扩增,通过不同信号的阳性液滴比率对目的基因进行定量。
基于数字PCR灵敏度和稳定性较高的优势,付伟等[20]于2015年开发了一种基于数字PCR的转基因成分定性筛查检测体系,通过以CaMV35s启动子以及NOS终止子作为扩增的目的片段,这两个筛选元件可以涵盖95%以上的国内批准的转基因品系(图4)。本实验后续进行了灵敏度、室内稳定性、实验室间稳定性、特异性及盲样检测的验证,验证结果表明,检测体系可以精准稳定的对质量百分比为0.1%的转基因样品实现筛查检测,绝对检测限可以低至单拷贝。相对于常规筛选检测体系,具有精准、稳定、结果呈现形式简单等优点。相对于常规基于数字PCR的转基因成分检测体系,本研究消除了样品前处理过程对检测体系的影响,可以更快速、更准确的实现转基因成分的初步筛查。

图4 基于数字PCR的转基因成分定性筛查检测体系[20]
相较于传统的PCR检测技术,数字PCR具有微滴数绝对定量的能力,因此数字PCR在转基因成分定量分析及鉴定方面也有了巨大的研究进展。朱鹏宇等[21]于2016年基于Fluidigm芯片式数字PCR平台,开发了一种不依赖于样品前处理步骤的转基因玉米成分绝对定量检测体系(图5),该研究前期验证了不同的影响因素对于体系的干扰,如SNP位点,样品前处理步骤等。在体系建立完成以后,又针对8种不同的转基因品系筛选出了最适合搭配的内标准基因的引物和探针,进而建立了针对8种不同的转基因品系的二重数字PCR绝对定量检测体系。该检测可以精准、快速的对未知样品中转基因成分进行定量检测,操作步骤少,结果稳定性高。
Chen等[22]采用双通道ddPCR定量检测方法对转基因水稻“汕优63”的成分进行分析,扩增“汕优63”的内源基因和菌株特异性序列,准确定量检测“汕优63”的成分。吴潇等[23]以转基因大豆中结构特异性基因为靶基因,利用ddPCR对其进行准确定量,并确定检测灵敏度高于国标中qPCR方法。Iwobi等[24]将ddPCR应用于选定的转基因食品和饲料样品,表现出了ddPCR方法的良好性能。
除了常规数字PCR转基因成分检测体系之外,有更强适用性的新型数字PCR的检测技术也得到了学者们的关注。牛晨启等[25]于2018年通过结合通用引物技术与数字PCR技术,开发了一种单通用引物多重数字PCR(Single universal primer-multiplexddPCR,SUP-M-ddPCR)转基因检测方法(图6)。研究中,反应体系添加了两种荧光探针,用来识别转基因序列的荧光探针(红色)和用来识别玉米内参序列的荧光探针(黄色)。体系中还同时有通用引物和5对修饰特异引物,即每一对特异引物的5′末端修饰一段相同的通用序列,来扩增P-35S、T-NOS、NPTII、PAT四种转基因筛选元件和玉米内参adh1基因。这个混合体系被分散在数字PCR的20 000个微滴中独立发生反应,在PCR过程前10个循环的第一阶段,反应体系中同时存在修饰特异引物和通用引物,修饰特异引物发挥作用,扩增出两端有通用引物的目标序列,第二阶段通用引物发挥巨大的作用,保证各个目标序列有统一的扩增效率。通用引物可以有效地减少多对引物之间的干扰。最后通过荧光信号的读取来对转基因成分进行检测和定量。该方法的优点是高通量,其中选取的4种转基因序列,覆盖了超过93%的目前已批准的转基因品系,可以在短时间内对大量样品进行初步筛查。
图5 基于Fluidigm芯片式数字PCR平台的转基因成分绝对定量检测体系[21]
3.2 转基因外源插入片段拷贝数鉴定
dPCR具有高精确度的特点,使用dPCR定量目标基因与参考基因的双重反应并计算它们的比值,得到目标基因拷贝数,在不同实验室质检具有很高的重复性,使其成为测量拷贝数的首选方法[26-28]。
dPCR可以实现复杂基因组拷贝数的准确测定。甘蔗是一种基因组较大且染色体具有高度多倍性和非整倍性的栽培种植物[29-30],没有已经准确测得拷贝数的基因,使用常规PCR很难检测。Yue等[31]通过ddPCR比较3种不同甘蔗品种中内源基因的绝对量,评估3种基因对拷贝数分析的适用性,推导出ATC基因可以成为未来基因拷贝数变异和Q208A和Q240A功能基因剂量研究的合适内源参考基因,并评估了ddPCR在高通量转基因测定中的应用(图7)。种质Bru1基因的拷贝数变异能够使甘蔗产生真菌病害甘蔗褐锈病的抗性,McCor[32]利用筛选的抗性甘蔗进行ddPCR拷贝数分析,说明ddPCR有望用于估计作物等位基因剂量,为育种等进一步研究提供有效信息。
2015 年,Félixurquídez等[33]基于双荧光通道数字PCR技术开发了一种鉴定转基因玉米中外源基因拷贝数的鉴定方法(图8),该方法以外源NOS终止子与玉米内源hmg基因为靶标序列,通过反应退火温度等反应条件的优化,可以对质量百分比在0.08%以上的转基因作物进行稳定的拷贝数定量。
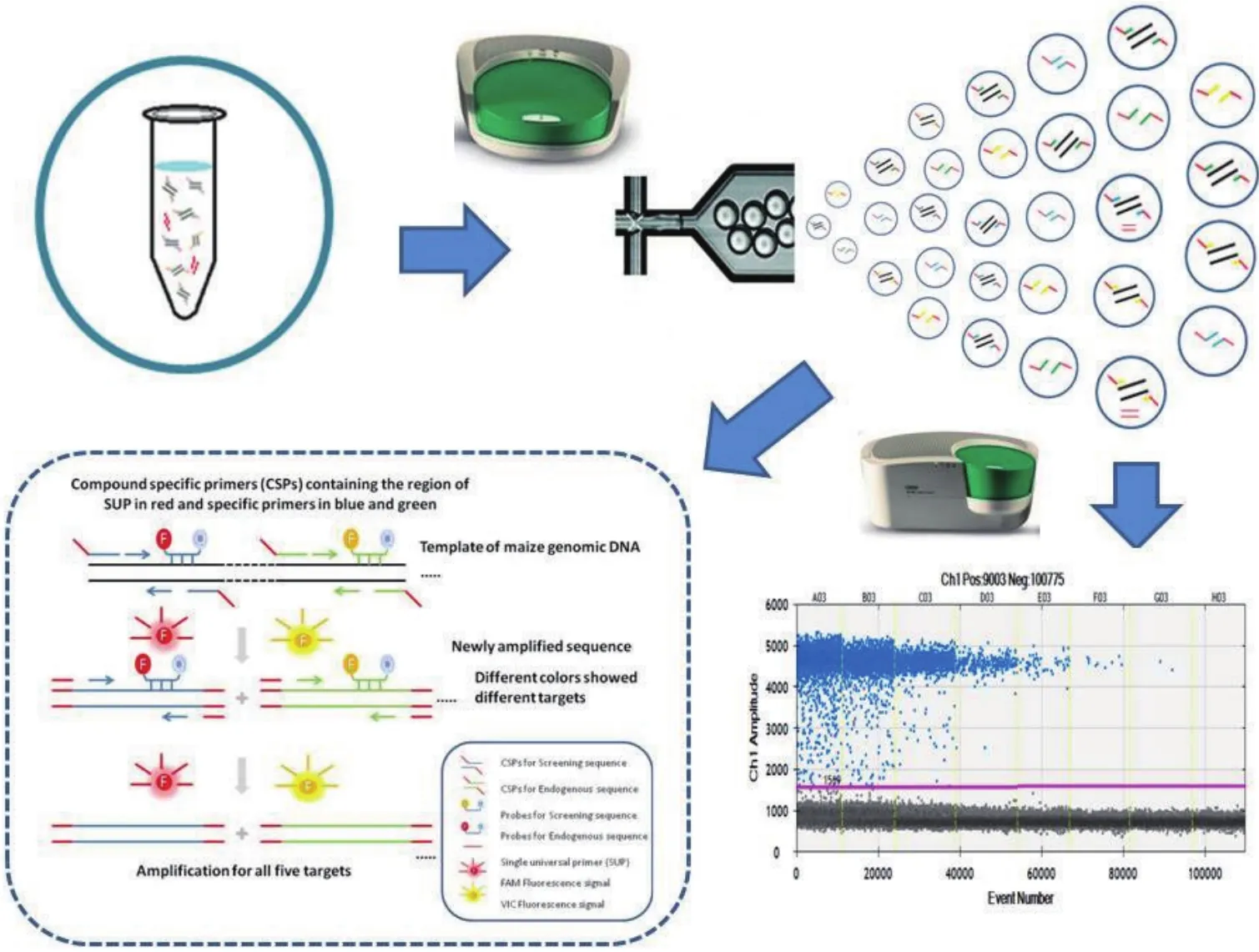
图6 单通用引物多重数字PCR转基因检测方法[25]
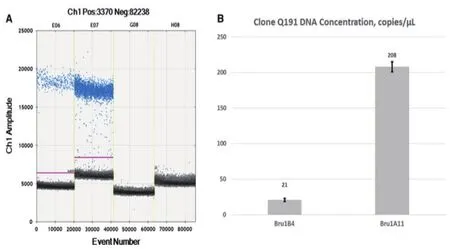
图7 抗性甘蔗中数字PCR等位基因分析结果图[31]
3.3 基因编辑
基因编辑通过序列特异性核酸酶诱导DNA产生切口或双链断裂,依赖于同源定向修复(Homology directed repair,HDR)或非同源末端连接(Nonhomologous end joining,NHEJ)实现定向修复[34-36]。ddPCR可以实现对复杂背景的分区,通过有限稀释,使得低丰度的目的序列能够被灵敏的检出,可以实现对含量较低的基因编辑事件的检测(图9)。数字PCR检测基因编辑的方法主要是将TaqMan PCR测定与ddPCR相结合,在靶标基因同一个扩增子内设计了一个通用引物对和编辑位点特异性TaqMan探针,与不同的荧光基团结合,据双重荧光信号判断编辑情况,区分野生型和诱变型DNA序列从而筛选诱变事件。
CRISPR-Cas9技术通常在修复编辑位点中存在有两种不同的修复机制HDR和NHEJ,其中前者相对于后者可以产生更加精准的基因片段修复结果。由于这两种修复方式几乎是存在于相同的编辑片段中的,因此目前常见基因编辑检测技术是无法对这两种修复方式的结果进行同时鉴定的。为了克服常用检测技术的缺陷,Yuichiro等[37]开发了一种可以系统、快速的鉴定HDR与NHEJ修复位点的数字PCR检测体系(图10)。这个研究中,Yuichiro以经过CRISPR-Cas9技术处理过的海拉细胞系(HeLa cells)为研究对象,通过在基因编辑片段中的NHEJ位点、HDR位点以及内源亲本位点为靶标序列设计探针,实现了通过一对引物的扩增就可以实现基因编辑位点检测以及NHEJ与HDR分型的目的。该研究可以稳定实现对1 000个野生型基因组中的单拷贝NHEJ/HDR位点的基因分型研究。
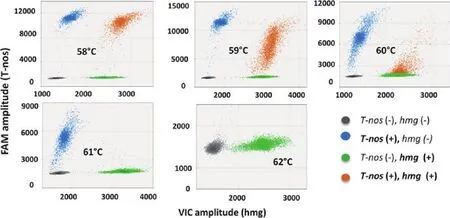
图8 双荧光通道数字PCR检测方法[33]
Findlay等[38]通过在原始序列Cas9酶切位点设计探针并采用ddPCR检测敲除诱导干细胞相关蛋白编码基因的改变,得到了敏感且特异的筛选基因编辑的方法,并通过ddPCR的阳性荧光信号比对实现了不同TALEN策略的效率评估。Mock等[39]以敲除了CCR5基因的人细胞为例,采用新一代测序(New generation sequencing,NGS)取得TALEN诱导的NHEJ的靶标序列,并针对这段被剪切过的序列设计NHEJ敏感的探针,使得产生NHEJ的靶标序列不产生荧光信号。由核酸酶介导的基因编辑(GEF-dPCR)利用两个不同标记的探针放置在基因编辑目标位点的一个扩增子内,以同时检测野生型和非同源末端连接影响的等位基因,通过dPCR结果来评估GEF-dPCR频率,并利用这种新设计的GEF-dPCR来测量CCR5基因中的TALEN介导的基因敲除,以及CCR2中预期的脱靶位点,以一个比较好的灵敏度和较大的线性范围对基因编辑进行量化。Miyaoka等[40]使用野生型和突变体等位基因的质粒模板混合物对数百个亲本细胞中含有单个点突变的细胞进行了检测,并发现ddPCR在野生型背景中的适应性可以在检测到0.1%的突变等位基因,灵敏度比单独TaqMan PCR高100倍。ddPCR在HEK293T细胞中使用PHOX2B和PKP2靶向的TALEN强力检测到TALEN诱导的点突变,并且比Surveyor测定更敏感。
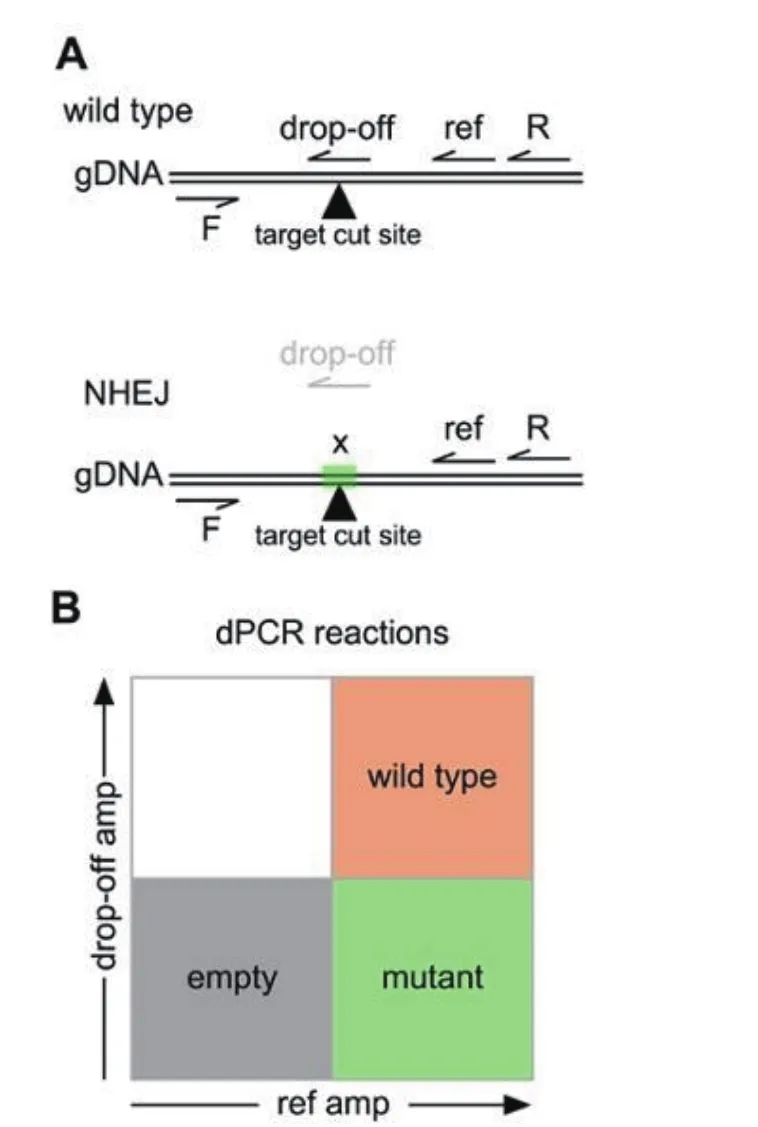
图9 探针检测方法[36]
总体来说,目前ddPCR检测基因编辑技术大多采用已知原始序列与模板序列设计HDR与NHEJ敏感的探针的方法,再经过背景不断稀释,实现对基因编辑产物有无以及编辑效率的判定。
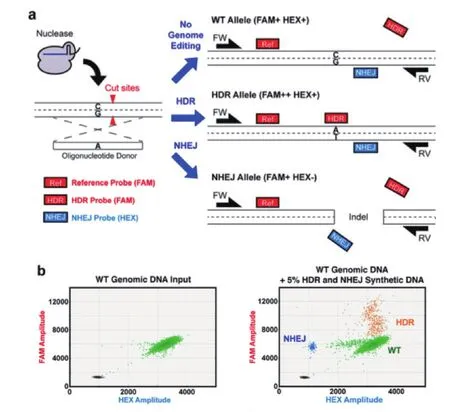
图10 数字PCR法NHEJ/HDR位点基因分型实验原理(A)及实验结果(B)[37]
4 数字PCR在诊断学功能核酸检测鉴定中的应用
dPCR具有高灵敏度和精确性的优势,除了痕量核酸分子检测、稀有突变等方向,在目前低丰度,背景来源复杂的医学领域里也有很广泛的应用。它可以提供研究人员探索复杂的遗传背景的可能性,发现并验证新的疾病关联,开辟分子诊断新的方向。
4.1 痕量核酸样品的检测与鉴定
临床肿瘤诊断主要以筛查患者外周血中特异突变的分子标志物为手段,其重点是血液中游离的肿瘤DNA的液体活检[41]。由于其具有背景复杂且高度片段化的特点,就要求采用灵敏度高、克服背景序列的检测方法。目前已有很多文献报道dPCR成功运用在各种癌基因的检测中,赵钊等[42]通过数字PCR检测非小细胞肺癌组织中的表皮生长因子受体EGFR基因T790M突变的差异。Feng等[43]也检测了79个肿瘤组织和配对血浆样本的血浆EGFR状态,并与Super ARMS的结果进行对比,验证两者结论具有一致性。Sedlak等[44]利用ddPCR对以染色体重组方式存在的人类疱疹病毒6型的人体血浆样本进行检测,灵敏度高达100%。
Beck等[45]证明抑制物DNA增加的量与移植排斥反应有关,同时推测了通过dPCR量化肿瘤衍生的细胞游离DNA(cfDNA)可能提供检测治疗功效的结果。应用定量分子测量对移植管理和监测病毒载量治疗实体瘤对提供全新的变化。布鲁菌病常规实验室诊断方法为菌株分离培养,但受环境影响大。郭瑛等[46]首次采用dPCR的方法对其进行探索性的技术检测,综合分析布鲁菌恢复期病例及dPCR检测血清布鲁菌DNA含量,评估dPCR取代血培养是有前景的但还需进一步验证。Deborah等[47]利用ddPCR对携艾滋病婴儿血液HIV核酸水平进行持续性检测,最终证实全球首例功能性治愈的艾滋病患儿。可以看出很多医学工作者逐步尝试用dPCR的方法替代原有检测方法实现更准确高效的诊断,目前商品化的ddPCR已被大量应用于肠病毒,巨细胞病毒,结核杆菌,耐甲氧西林金黄色葡萄球菌等临床领域[48-50]。
4.2 拷贝数变异的检测与鉴定
数字PCR可以绝对定量的特点,使其在鉴定拷贝数变异方面相较于常规的PCR技术具有明显的优势,现阶段,数字PCR已经在肿瘤相关基因拷贝数变异鉴定方面取得了广泛的应用。
CNV通常是指DNA部分(包括基因)的重复与缺失,Benjamin等[51]首次应用ddPCR在高通量样品条件下对二代测序技术进行验证,此后随着与NGS等方法的联用来进行CNV位点的验证越来越得到认可。目前研究表明剂量的变化会影响基因的表达[52]与表型[53-54]。随着拷贝数的增加,量化剂量效应变得具有挑战性,更具准确性的dPCR被采用。Maria等[55]研究唾液淀粉酶基因的高度多态拷贝数变异体,他们利用ddPCR加转录组学的方法分析用于检测潜在的基因剂量效应(图11)。发现AMY1拷贝数与淀粉酶的量呈显著正相关,对降低肥胖风险有显著影响[56]。也说明了其他形式的DNA变异包括拷贝数变异可能解释遗传性的缺失。
Miotke等[57]开发了一种单色荧光dPCR的方法,使用非特异性双链结合染料EvaGreen(EG),通过操纵目标和参照扩增子的长度实现区分它们的荧光信号并独立量化(图12)。Miotke等通过检查原癌基因FLT3中的拷贝数和BRAF中常见的V600Epoint突变来证明这种方法的有效性。扩增子的长度改变导致双链DNA存在的碱基对改变。由于EG染料的荧光幅度与dsDNA存在量成正比,具有较长扩增子的微滴比具有较短扩增子的微滴荧光更明亮。这种检测方法可以减小投入,提供了商业推广的可能性。
除了这些比较常见的方法,还有学者开发了具有针对性的测定方法。Taly等[58]开发了一种不同浓度的相同荧光基团反应测定7种突变的多重方法(图13),结合ddPCR可以实现简单的格式来筛选多个基因座的可能性。可以通过改变TaqMan探针的浓度和性质来调节终点荧光强度,这使得可以识别和计数含有每个独特可扩增靶标的液滴。
5 数字PCR在微生物检测方向的应用
根据世界卫生组织的估计,全世界每年的食源性疾病患者大部分都是由致病微生物引起的,从原材料加工到流通贮藏各个环节都有可能感染,相比于常规的微生物检测需要繁复的增菌、分离和生物学鉴定,PCR技术更加快速灵敏且特异性强。ddPCR定量检测食源性微生物是目前发展的一个方向。张永江等[59]根据马铃薯S病毒的外壳蛋白基因保守序列设计引物探针;陈嘉茵等[60]采用逆转录微滴式数字PCR可以灵敏地检测出受污染食品中低病毒含量。Rothrock等[61]用ddPCR对商业家禽处理水样进行沙门氏菌检测;Bian等[62]对单核细胞增生李斯特菌进行检测,检测结果表明方法最低检测下限为10 CFU/mL。ddPCR方法在实际应用中有很好的特异性与灵敏度,在食源性微生物中有很大发展空间。qPCR在炭疽杆菌质粒拷贝数检测中差异很大[63-64]。Straub等[65]采用 ddPCR 对炭疽杆菌质粒pXO1和pXO2进行了拷贝数测定。将qPCR与ddPCR的实验结果进行对比发现dPCR结果更接近于直接测序数据。
ddPCR还可以实现自然界中多个细菌多个基因的鉴定与分类。Ottesen等[66]用编码参与白蚁和肠道菌群共生的关键酶基因作为实验钩,发现几个未知核糖体RNA并鉴定了复杂环境中携带目的基因片段的菌群,Hua等[67]利用ddPCR分析土壤中黄曲霉毒素和无毒黄曲霉菌混合物的群体比率,由于黄曲霉无毒素菌株与产黄曲霉菌菌株具有竞争关系,可用于后续生物防治。
6 数字PCR在非核酸靶标物质中的检测中的应用
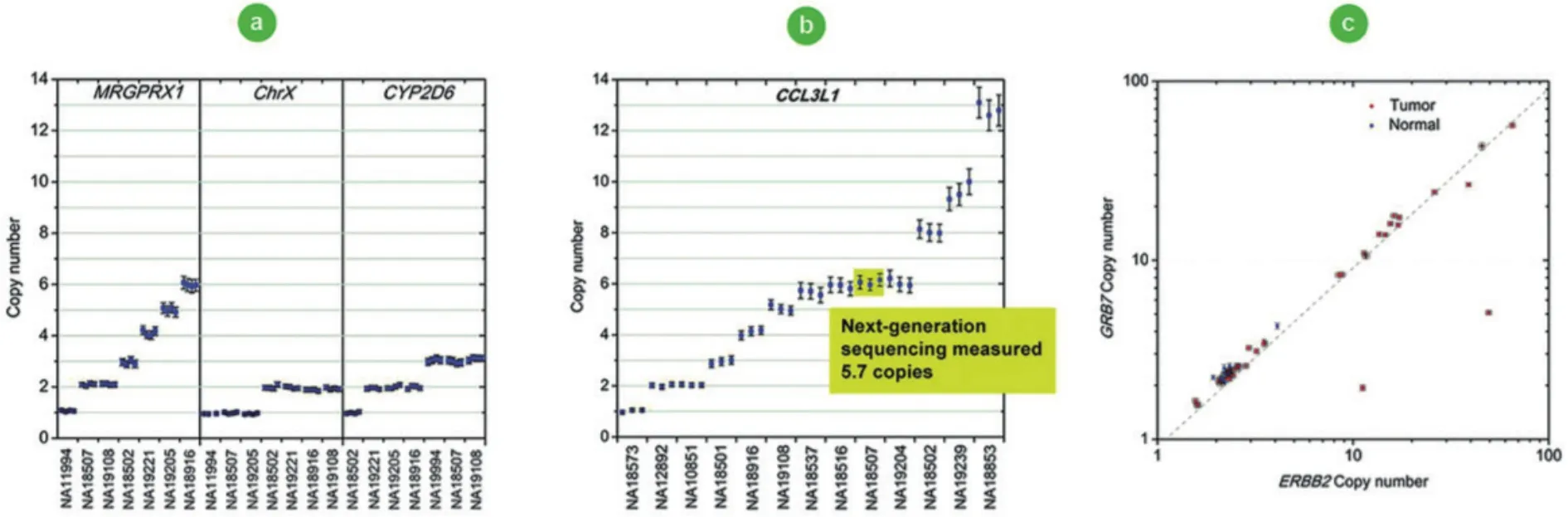
图11 数字PCR检测拷贝数[55]
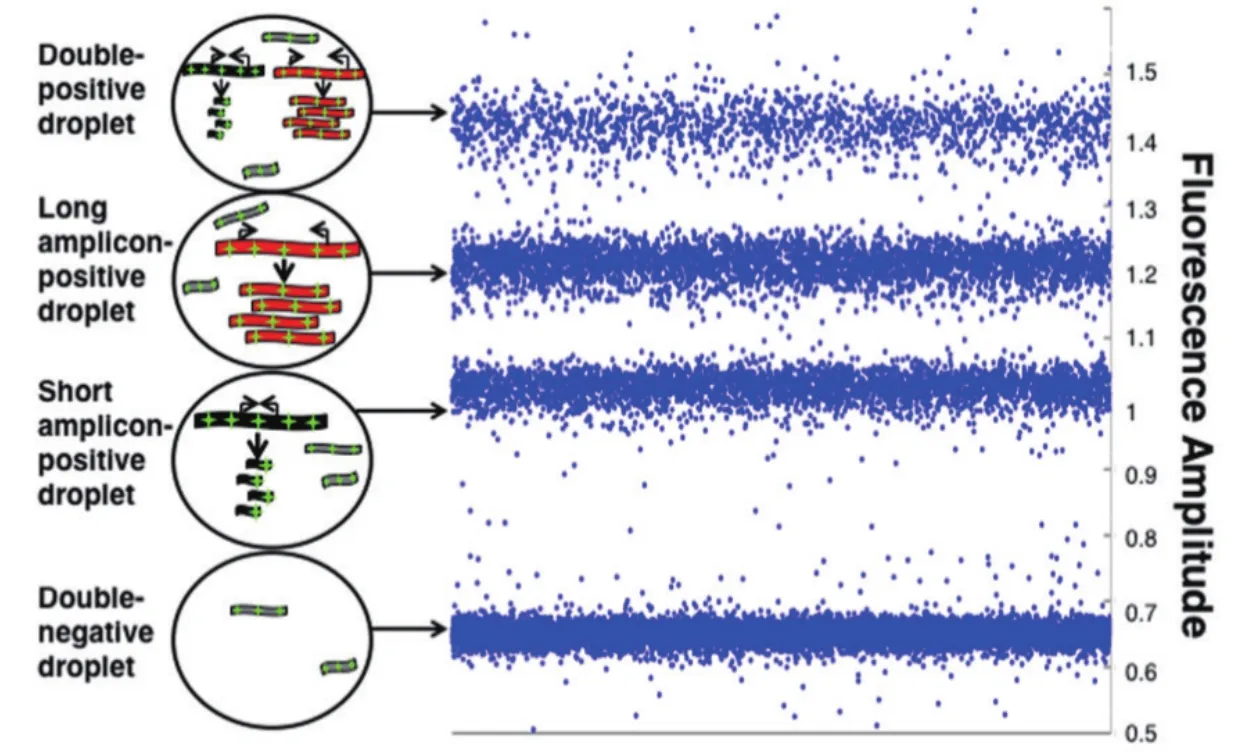
图12 单色荧光数字PCR检测方法[57]
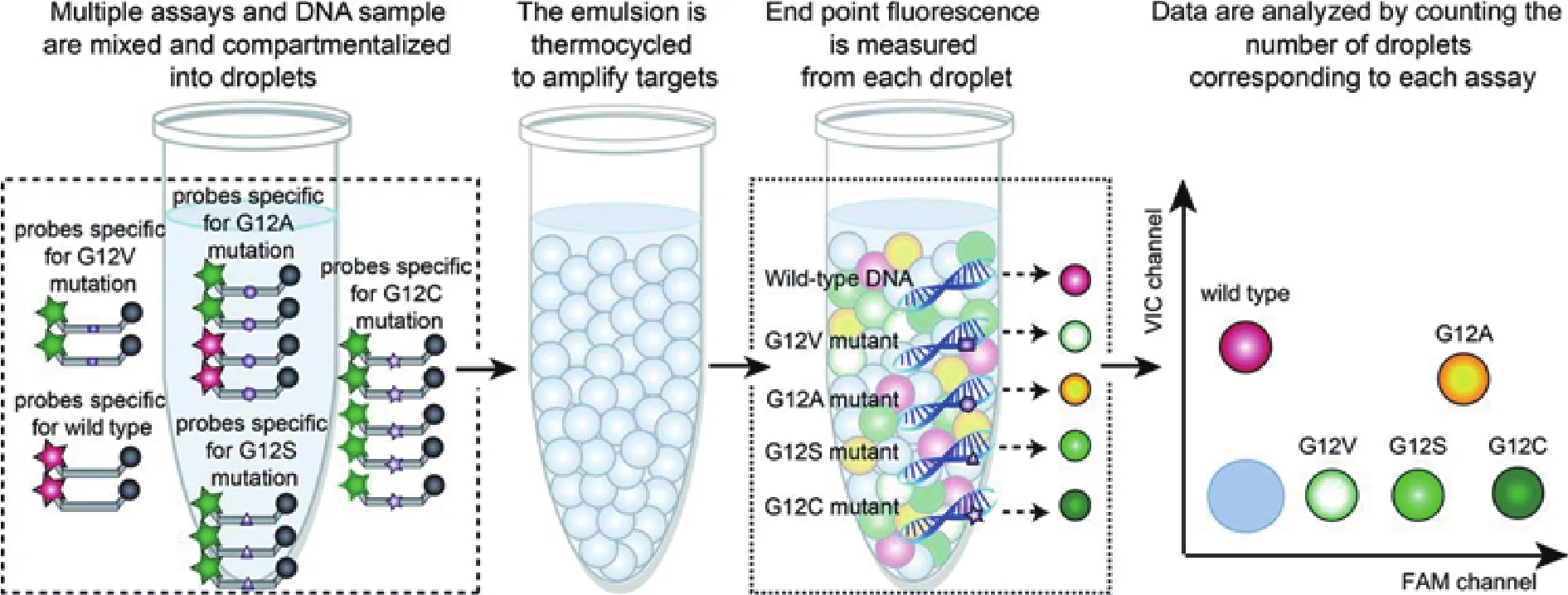
图13 单色荧光多重检测方法[58]
生物传感器是一类可以实现不同种类信号之间转化的装置,其中核酸传感器是一类可以将非核酸信号转化为核酸信号的核酸分子。常见的核酸传感器包括重金属核酶传感器、核酸适配体生物传感器、电化学生物传感器等。以重金属核酸传感器为例,传统的重金属检测技术通常是基于ICP-MS等大型仪器的,随着便携性更强的重金属传感器的开发,针对不同重金属离子的核酸检测技术已经越来越得到学者们的关注。
许文涛团队[68-69]结合数字PCR技术与重金属传感器,开发了针对铜离子的开型(Turn-on)生物传感器检测技术以及汞离子的闭型(Turn-off)生物传感器检测体系。该研究以铜离子(Cu2+)和汞离子(Hg2+)为研究对象,通过体系优化,实现了生物传感器步骤从重金属离子信号到核酸信号最为高效的转化(图14)。后续通过对核酸信号进行数字PCR的绝对定量鉴定以及信号转化率计算,实现了对两种重金属离子的绝对定量。对于两种重金属离子,本检测体系的定量检测限可以达到500 fmol和40 fmol,定性检测限达到50 fmol与10 fmol,线性范围均超过3个数量级。该检测体系的检测限以及线性范围表明该检测体系可以满足绝大多数样品中两种重金属离子的检测需要。
另一方面,为满足汞离子和银离子的逻辑门检测方法的需要,程楠等[70]首次通过数字PCR产生的数字信号建立了一系列的绝对定量逻辑门检测监测体系(图15)。由于汞离子和银离子特殊的生物化学性质,依赖T-Hg-T和C-Ag-C的错配碱基配对,实现了重金属信号向核酸信号的转化。通过进一步的体系优化,实现了对重金属离子的绝对定量检测和不同类型逻辑门的设计(YES门、AND门和OR门)。

图14 基于生物传感器和数字PCR的铜离子以及汞离子绝对定量检测体系原理图
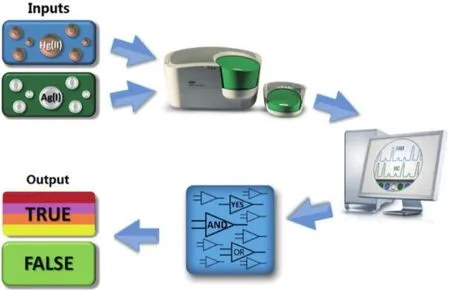
图15 数字PCR-逻辑门检测体系基础反应原理[71]
7 小结
数字PCR技术的广泛应用逐渐显现出一些不足之处。首先,相对于常规检测技术来说,由于数字PCR荧光通道的局限性导致其检测通量不会很高,这决定了在多样品多靶标的筛查中,数字PCR技术通常具有较高的工作量;另外,数字PCR较为昂贵的实验成本也同样制约了数字PCR的发展;而且,由于数字PCR平台商业化还没有特别成熟,所以数字PCR平台的自动化操作还没有得到学者们的广泛关注,同时数字PCR平台的标准化检测技术开发的滞后也制约了数字PCR技术在各领域的进一步发展;另一方面,由于现阶段数字PCR内部的平台众多,各个平台的数据分析处理方式不尽相同,所以如何对不同数字PCR平台之间的数据进行规范的协同分析也是数字PCR发展的重点。不过由于数字PCR较于常规检测技术不可替代的优势,尤其是在背景复杂的样品方面[71],其不断发展也将对功能核酸检测领域产生深远影响,在分子生物学和医学等基础研究和应用方面发挥更大的作用。
猜你喜欢
学与玩(2022年10期)2022-11-23 08:32:00
中华诗词(2022年9期)2022-07-29 08:33:50
中国慈善家(2022年3期)2022-06-14 22:21:55
今日农业(2022年3期)2022-06-05 07:12:08
快乐语文(2021年34期)2022-01-18 06:04:14
河北医学(2021年10期)2021-10-27 00:37:14
中国(俄文)(2020年8期)2020-11-23 03:37:13
中国临床医学影像杂志(2019年6期)2019-08-27 02:59:50
创新科技(2015年1期)2015-12-24 06:23:21
发明与创新(2015年25期)2015-02-27 10:39:16