
肿瘤微卫星不稳定检测方法综述①
2018-10-24 11:05李晓东何小雨李瑞琳牛北方
计算机系统应用 2018年10期
陈 玮, 赵 丹, 李晓东, 何小雨, 李瑞琳, 牛北方,3
1(中国科学院 计算机网络信息中心, 北京 100190)
2(中国科学院大学 计算机与控制学院, 北京 100049)
3(贵州大学 医学院, 贵阳 550025)
微卫星, 即短串联重复序列, 是广泛分布在真核生物基因组中的(原核生物基因组中也有少量分布), 以1-6bp为一个重复单元, 重复次数不超过60次的DNA序列. 人类基因组中有数以万计的微卫星位点,这些微卫星位点近似均匀地分布在各个染色体上, 所有的微卫星序列约占整个基因组的3%. 微卫星按照重复单元的大小可分为单核苷酸、二核苷酸、三核苷酸、四核苷酸、五核苷酸、六核苷酸重复;按照重复序列的结构可分为简单重复(由单一重复单元构成)和复合重复(由重复单元不同的多个重复序列构成). 与DNA中的其他区域相比, 微卫星区域具有较高的突变率[1]. 其高突变率的直接表现是高度的多态性[2], 即不同个体之间或正常组织与肿瘤组织之间, 微卫星位点重复单元的重复次数存在差异. 由于微卫星位点的分布广泛性及高度多态性, 微卫星常用于个体鉴定、连锁图谱的绘制以及肿瘤发生机制的研究.
微卫星不稳定(Micro Satellite Instability, MSI), 是指微卫星位点重复单元的重复次数出现波动的现象,即重复单元的插入与删除. 现普遍认为这种现象是由DNA在复制过程中出现“链滑”(strand slippage)引起的. DNA在复制过程中, DNA聚合酶沿模板链滑动,子链与模板链会发生局部分离和重新配对. 在重新配对的过程中, 子链与模版链发生错配, 就会导致一个或几个重复单元形成凸环. 一般情况下, 这种错误会被DNA的错配修复系统(Mismatch Repair, MMR)修复,然而, 当MMR中的相关基因由于启动子超甲基化或基因突变等原因出现故障, DNA复制错误无法被修复,一些微卫星位点重复单元的重复次数发生波动, 进而发生微卫星不稳定[3]. 不同的微卫星位点稳定性不同.微卫星重复单元的大小、重复单元的碱基组成、重复序列的结构及重复次数等都会在一定程度上影响位点的稳定性. 根据微卫星不稳定的程度, 可以将MSI分为MSI-H (MicroSatellite Instability High), MSI-L(MicroSatellite Instability Low)以及MSS(MicroSatellite Stable).
1993年, Aaltonen等人首次在家族性遗传性结直肠癌(Hereditary Non-Polyposis Colorectal Cancer,HNPCC)中发现高频率的MSI[4]. 微卫星不稳定在大约15%的结直肠癌以及90%的林奇综合症(HNPCC,又称Lynch Syndrome)中起决定作用[5]. 近年来的研究表明, MSI对林奇综合症以及结直肠癌的诊断、预后以及化疗敏感性有重要的意义. 除了结直肠癌, 研究人员也相继在子宫内膜癌、卵巢癌[6]、胃癌以及乳腺癌[7]等疾病中发现MSI. MSI作为肿瘤遗传不稳定的敏感指标, 其检测对于肿瘤的早期诊断、预后判断、化疗敏感性判断以及高危人群的圈定等具有重要意义. 已有不少研究发现MSI-H的肿瘤患者相对于MSS的肿瘤患者有更好的预后[8,9], 同时MSI-H肿瘤患者对不同化疗方法的敏感性也表现出差异[10].
目前, 临床上主要采用MSI-PCR以及MMRIHC的方法进行微卫星不稳定的检测. 然而, 近年来,随着下一代测序技术(Next-Generation Sequencing technology, NGS)的飞速发展, 测序价格以超越摩尔定律的速度急速下降, 测序速度也大幅提升, 这使得方便快捷地获取测序数据成为可能. 目前已有多个通过分析测序数据来检测微卫星不稳定的软件方法.
下文将从基于生物学实验的方法和基于计算的方法两个角度来介绍现有的微卫星不稳定的检测方法.
1 基于生物学实验的方法
当前临床上主要采用聚合酶链式反应(Polymerase Chain Reaction, PCR)或免疫组织化学(Immuno Histo Chemistry, IHC)染色的方法检测患者的MSI状态.
MSI-PCR[11]通过对肿瘤组织和正常组织中选定的微卫星位点进行PCR扩增及凝胶电泳, 通过比较两组电泳结果的差异来确定MSI的状态. 然而人类基因组中有数以万计的微卫星位点, 不同的位点对于检测MSI的敏感性和准确性也各不相同. 为了标准化MSI的检测, NCI (National Cancer Institute)于1997年推荐了Bethesda指南[12], 该指南推荐了两个单核苷酸位点(BAT-25, BAT-26)以及三个二核苷酸位点(D2S123, D5S346 和D17S250)作为检测MSI的微卫星标记, 检测结果中有两个及以上位点出现不稳定为MSI-H, 一个位点出现不稳定为MSI-L, 没有位点出现不稳定为MSS. 鉴于二核苷酸位点在对MMR故障的肿瘤患者的MSI检测中, 敏感性和准确性不及单核苷酸位点, NCI又于2004年对Bethesda指南进行了修订[13].与此同时, Bacher等人[14]通过对266个微卫星位点(其中包括单核苷酸、二核苷酸、四核苷酸以及五核苷酸微卫星位点)检测的敏感性及准确性进行评估, 提出了Promega分析系统, 该系统使用五个单核苷酸微卫星位点(BAT-25, BAT-26, NR-21, NR-24和MONO-27)检测MSI, 并使用两个五核苷酸微卫星位点(Penta C和Penta D)标识样本.
与MSI-PCR不同, MMR IHC通过检测MMR蛋白(MLH1、MSH2、MSH6和PMS2)的表达来确定MMR系统是否发生故障, 进而判断MSI的状态. 然而并不能用MMR IHC完全替代MSI PCR, 因为在确定为MSI-H的肿瘤中, 有5%的肿瘤, 四种蛋白都表达,使用MMR IHC无法将其识别.
2 基于计算的方法
目前, 已有多个通过分析高通量测序数据检测微卫星不稳定的方法及软件. 从模型的角度可以将这些方法分为基于一般统计模型的方法和基于机器学习模型的方法. 其中, 基于统计的方法, 首先选取一个可以反映微卫星不稳定特点的指标, 然后在一组给定的样本上(MSI的临床检测结果已知), 确定该指标与临床检测结果的一致性及分类阈值. 基于机器学习的方法,主要通过特征提取、特征选择及分类器训练的方法进行MSI状态的预测. 不论是统计方法中的指标还是机器学习方法中的特征, 其选择的主要依据是微卫星不稳定这一现象以及其背后的产生机制. 其中, 现象, 即测序数据中表现出的微卫星位点重复单元重复次数的波动, 其本质上是碱基的插入与删除;产生机制, 即DNA错配修复系统相关基因启动子超甲基化或发生突变使得这些基因无法表达, 进而影响到错配修复系统的功能. 因此, 基于计算的方法一般是通过对测序数据、超甲基化数据、突变数据以及基因表达数据进行分析, 确定MSI状态的.
从样本的角度可以将这些方法分为基于配对的肿瘤-正常样本的方法和仅基于肿瘤样本的方法. 第二种方法在缺乏与肿瘤样本配对的正常样本的情况下, 可以有效解决MSI的检测问题.
表1从以上两个维度对现有的方法进行了分类.

表1 MSI检测方法分类
以下将从模型的角度分类介绍各个方法.
2.1 基于一般统计模型的MSI检测方法
目前, 主要有以下四种基于一般统计模型的MSI检测方法, 这些方法均是通过对测序数据的分析,从微卫星位点重复单元重复次数波动的角度出发, 解决这一问题的.
(1) 基于Indel的MSI检测方法[18]
MSI中发生的重复单元的插入与删除从本质上是小片段碱基的插入与删除, 即Indel. Lu等人正是从这个角度出发, 将MSI的判定问题转化为了微卫星区域的Indel变化问题.
对于每个样本, 首先进行Indel识别, 其次对获得的Indel进行过滤并保留位于微卫星区域的Indel. 通过在一组样本(MSI临床检测结果已知)上对PI、PD以及PI/PD作为MSI判别指标进行t检验评估(其中PI表示微卫星区域insertion占所有insertion的比例, PD表示微卫星区域deletion占所有deletion的比例, PI/PD为二者的比率), 选择了PI/PD作为样本的MSI判别指标. MSI-H的样本在该指标上的取值显著低于MSS的样本.
Lu等人仅提供了上述方法的工作流程并通过实际的数据验证了该方法的有效性, 并没有开发出相应的软件工具.
(2) mSINGS[19]
mSINGS首先判断每个微卫星位点的稳定性, 进一步根据不稳定的微卫星位点的比例来判断样本的MSI状态. 对于每个微卫星位点, mSINGS试图找到一个指标来量化其稳定程度, 并基于一组MSS样本建立各微卫星位点该指标的参考值, 对于给定样本的某个微卫星位点, 若该指标取值超出参考范围, 则认为该微卫星位点不稳定. 通过这种方式, mSINGS解决了仅有肿瘤样本情况下MSI的判定问题. 具体方法如下:
1) 对于任一微卫星位点, 以其等位基因的个数作为衡量该位点是否稳定的指标, 计算一组MSS样本上,该位点等位基因个数的平均值作为参考值. 具体计算方法如下:
① 仅选择在该位点测序深度大于等于30的MSS样本参与计算;
② 对每个符合条件的样本, 计算该位点等位基因的分布信息, 如表2所示;
③ 对每个符合条件的样本, 规范化其等位基因的支持reads数:规范化的支持reads数=支持reads数/最大支持reads数;
④ 对每个符合条件的样本, 过滤掉规范化的支持reads数小于5%的等位基因, 以剩余的等位基因数作为该样本该位点的等位基因数;
⑤ 计算符合条件的样本该位点等位基因数的平均值(该微卫星位点的参考值)及方差.
2)对于给定样本, 采用与1)相同的处理方式, 对比1) 中建立的参考值, 根据3σ法则判断其各微卫星位点的稳定性;
3)计算不稳定微卫星位点的比例以判定样本的MSI状态.
从上述mSINGS的方法介绍可以看出, 各微卫星位点稳定性指标的参考值是影响mSINGS准确性的重要因素, 而参考值的计算依赖于合理地选择一组MSS样本. 为了保证判别的准确率, 用于参考的MSS样本与待检测的样本应该具有较好的一致性, 如测序、癌种方面的一致性. 在实际使用中, 常常需要自行建立参考值.

表2 等位基因分布信息
(3) MSIsensor[15]
与mSINGS相似, MSIsensor也是通过分别判断每个微卫星位点的稳定性, 然后以不稳定微卫星位点的比例作为MSI得分. 不同的是, MSIsensor需要基于配对的肿瘤-正常样本进行MSI的判定. 首先, 对于在肿瘤和正常样本中测序深度均大于等于20的微卫星位点, 计算其等位基因的分布信息;其次, 通过卡方检验比较肿瘤和正常样本的相同微卫星位点的等位基因分布, 若显著不同, 则认为该微卫星位点不稳定;最后统计不稳定位点的比例, 若该比例超过阈值, 则判定为MSI-H, 其中, 阈值是通过该指标在一组样本上(包括MSI-H和MSS的样本)的累积分布确定的.
(4) MANTIS[16]
类似于MSIsensor, MANTIS也获得了肿瘤-正常配对样本在每个微卫星位点的等位基因分布信息;与MSIsensor不同的是, 对于每个微卫星位点, MANTIS把上述两组数据看作两个向量, 定义这两个向量的L1范数为样本中该位点的稳定程度, 对所有位点的L1范数求平均值即为样本的MSI得分. 具体方法如下:
对于每个微卫星位点,
1)仅保留读长、测序质量符合要求的比对到该位点的reads;
2)分别计算配对的肿瘤-正常样本中该位点的等位基因分布;
3)根据3σ法则, 过滤掉配对的肿瘤-正常样本在该位点支持reads不足的等位基因;
4)经过上述处理, 仅保留在配对的肿瘤-正常样本中支持reads总数(该位点的测序深度)均超过一定阈值的微卫星位点.
5)分别规范化肿瘤-正常样本该位点等位基因的支持reads数:规范化的支持reads数=支持reads数/该位点的总支持reads数;
6)根据规范化后的支持reads数, 计算配对的肿瘤-正常样本中该微卫星位点等位基因分布的L1范数;
7)以所有位点L1范数的平均值作为样本的MSI得分.
MANTIS对参与计算的数据进行了相对严格的质量控制, 如上述流程中的1)、3)及4)步骤. 由于测序过程中总会产生误差和错误, 通过质量控制, 仅使用符合要求的数据参与计算, 可以在一定程度上提高后续分析的准确性.
上述基于一般统计模型的MSI检测方法通过设计一个MSI判定指标, 在一组样本上, 使用累积分布等方式, 确定该指标的阈值, 实现对MSI状态的检测.MANTIS一文从MSI判定的准确性及计算资源使用两个方面对mSINGS、MSIsensor以及MANTIS三种方法进行了评估, 阈值、用于分析的微卫星位点的数量以及癌种都会影响软件的准确性. 尽管在敏感度和特异度方面有细微差异, 三个软件工具均可以准确的检测样本的MSI状态. 然而, 不同于mSINGS和MANTIS, MSIsensor没有对等位基因分布中的支持reads数进行规范化以及质控, 在配对的肿瘤-正常样本测序深度不同的情况下, 可能出现假阳性的结果.
2.2 基于机器学习模型的MSI检测方法
目前, 基于机器学习模型的MSI检测方法主要有以下三种. 特征和算法是机器学习的重要组成, 以下将从这两个方面介绍各个方法. 关于每个特征的提取方式不在此赘述.
(1) MSIseq[20]
发生微卫星不稳定的样本其单核苷酸替代(Single Nucleotide Substitution, SNS)率以及小片段碱基的插入与删除(Indel)比率都会发生变化, MSIseq主要是从基因变异这一角度出发选取特征的. 备选特征如表3所示.

表3 MSIseq备选特征
在这些特征的基础上, MSIseq使用五折交叉验证分别基于LR、决策树、随机森林、朴素贝叶斯算法训练了分类器并评估了性能, 最终选择基于决策树算法的分类器, 该分类器仅使用了S.ind这一个特征.
由于MSIseq提取的特征并不依赖于配对的肿瘤-正常样本, 因此这一方法适用于仅有肿瘤样本的情况.
(2) MOSAIC[17]
MOSAIC是基于对每个微卫星位点稳定性的判断设计特征的. 除了与各微卫星位点稳定性相关的特征外, 还增加了通过在一组样本上对所有微卫星位点的稳定性进行分析后发现的显著不稳定的微卫星位点信息, 备选特征如表4所示.

表4 MOSAIC备选特征
其中, 微卫星位点不稳定性的确定采用高敏感度的方法, 数据处理过程与mSINGS相同, 不同点在于微卫星位点不稳定的判定不再依据3σ原则, 而是对于任意微卫星位点, 若肿瘤样本相对于配对的正常样本, 在该位点出现新增的等位基因, 即认为该位点不稳定.
MOSAIC分别基于决策树和随机森林算法训练了模型, 最终选择了基于决策树算法的分类器, 该分类器仅使用了peak_avg以及defsite两个特征 .
MOSAIC选择的特征依赖于配对的肿瘤-正常样本, 因此仅适用于有配对样本的情况.
(3) MIRMMR[21]
与其他方法不同, MIRMMR的特征选择主要依据MSI的发生机制. 使用了与DNA错配修复系统相关的35个基因的点突变率、甲基化水平以及CADD[22]得分作为备选特征, 基于LR算法构造了分类器. 相比于MSIseq及MOSAIC, MIRMMR提供了更多的建模方法, 包括univariate、stepwise与penalized三种模式.其中univariate用于单变量的逻辑回归建模, 可以用于比较各特征用于MSI判定的准确性;stepwise模式用于自动化的特征选择, 从备选特征中, 选择最优的特征集训练模型;penalized模式在模型中增加了惩罚项用于防止过拟合. MIRMMR默认使用了penalized模式基于676个样本训练了模型. MIRMMR使用的特征不依赖于配对的肿瘤-正常样本, 因此可适用于仅有肿瘤样本的情况.
2.3 各方法的比较
针对上述提到的七种用于微卫星不稳定检测的计算方法, 从适用范围、MSI指标、测试数据集以及软件特性等方面进行了比较, 具体如表5所示. 其中“—”表示无相关信息, WES (Whole Exome Sequencing)表示全外显子组测序.
对于MSI的检测, 表5中每个方法的输出既可以是连续的MSI指标也可以是确定的分类. 其中基于一般统计模型的MSI检测方法, 可以在连续的MSI指标基础上, 根据阈值对样本分类;基于机器学习模型的MSI检测方法, 可以预测类别也可以输出类别的概率.
从软件易用性的角度分析, MSIsensor和MANTIS由于可以直接对BAM文件进行分析因此使用最为方便;而mSINGS在对样本进行分析之前, 需要足够的MSS样本建立参考值, 对样本量有一定要求,给使用带来了一定程度上的不便;基于机器学习模型的方法, 虽然可以直接使用模型进行预测, 硬件资源使用少, 速度快, 但是特征的提取依然是一个复杂低效的过程.
不论是基于一般统计模型的方法还是基于机器学习模型的方法, 要准确地检测MSI都离不开数据的支持, 测试数据集的大小和包含的癌种都会在一定程度上影响分类的准确性.
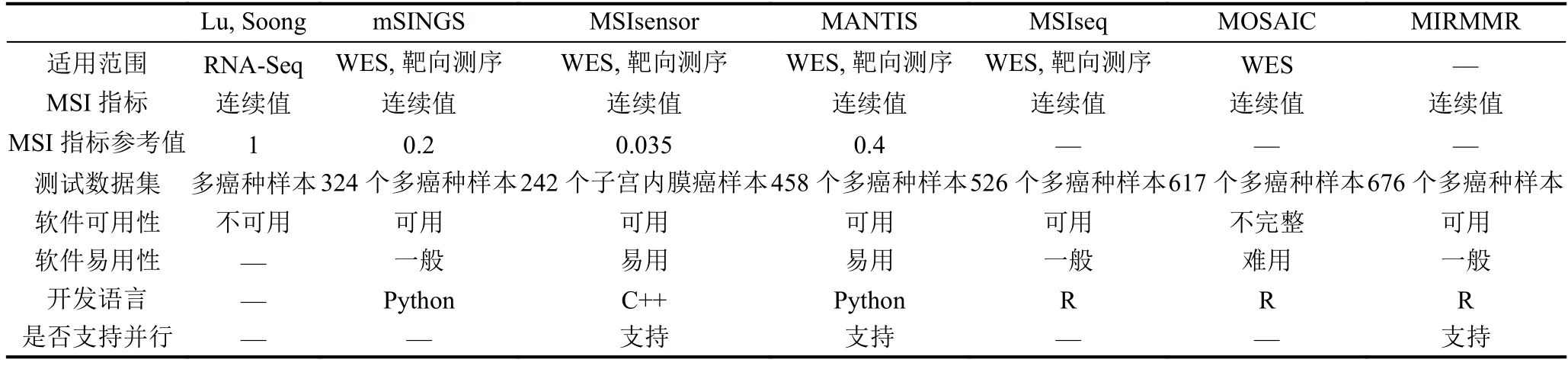
表5 基于计算的各方法比较
3 讨论
随着测序成本的下降和测序速度的提升, 计算方法相对于生物学实验方法的优势也越来越突出. 相比于计算方法, 通过生物学实验方法检测MSI有以下几个方面的不足. 首先, 需要耗费一定的时间和人力;其次, 结果的准确性依赖于分析人员的肉眼判断;再者,微卫星标记和MMR蛋白都有其局限性. 对于微卫星标记, 实验中选择的数量有限, 存在组织(肿瘤)特异性[23],无法准确地在多种肿瘤中检测MSI状态;对于MMR蛋白, 由于MMR可能不是引起MSI的唯一原因[24], 以及MMR自身的复杂性, 使用MMR蛋白的表达来间接判断MSI状态也存在局限性.
计算方法利用测序数据, 从MSI的表现及产生机制的层面, 可以对样本的MSI状态作出全面的评估. 相比于生物学实验方法, 计算方法的众多优势使其可能在未来用于微卫星不稳定的临床检测. 在这个过程中,还需要考虑以下方面的问题. 首先, 数据支持. 不论是基于一般统计模型的方法还是基于机器学习模型的方法, 要确定合适的阈值或提高分类器的准确性都需要大量数据的支持. 其次, 软件易用性. 软件要易于安装,其使用应该在最大程度上实现自动化同时运行时间需要在可接受的范围内.
MSKCC(Memorial Sloan Kettering Cancer Center)最近的一项研究[25]使用MSIsensor对12 288例实体癌病人的靶向测序数据进行分析, 判定MSI状态, 并用MSI-PCR/MMR-IHC进行了验证. 实验证明, 基于大规模靶向测序数据, 通过MSIsensor预测病人的MSI状态具有高的可信度. 对于MMR故障的样本, 相比于当前普遍使用的MSI-PCR方法,MSIsensor具有更高的敏感性. 根据2.3节的分析,MSIsensor使用C++语言开发, 安装及使用十分便利,同时支持并行计算, 运行速度快, 方便临床应用. 此项研究在一定程度上为该软件工具的临床应用提供了支持.
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
烟台大学学报(自然科学与工程版)(2022年3期)2022-06-30
肝博士(2022年3期)2022-06-30
水产科学(2022年2期)2022-03-20
南京师范大学学报(工程技术版)(2021年2期)2021-10-20
智慧健康(2021年17期)2021-07-30
四川动物(2020年1期)2020-02-27
新课程·下旬(2018年9期)2018-11-14
青少年科技博览(中学版)(2015年10期)2015-01-11
