
非均相固化体系对复合材料树脂微观力学均匀性的影响
2018-10-18 08:43郭妙才洪旭辉李亚锋
材料工程 2018年10期
郭妙才,洪旭辉,李亚锋
(1 中航工业复合材料技术中心,北京 101300;2 中国航发北京航空材料研究院 先进复合材料重点实验室,北京 100095)
连续纤维增强复合材料具有优异的比强度、比刚度,作为结构材料在飞机、船舶、汽车等领域中的应用越来越广泛[1-2],以环氧树脂为代表的热固性树脂在纤维增强复合材料中大量使用,其强度、韧性、缺陷特征、界面等因素决定复合材料整体的性能[1,3]。
使用潜伏型固化剂的环氧树脂是将常温时具有不溶性的固化剂颗粒加入并分散到树脂中的一类重要非均相固化树脂体系。这类树脂具有长的储存期,在固化温度下又可以快速溶解引发体系聚合,如以双氰胺为代表的潜伏型固化剂在预浸料中广泛使用[4-6]。然而这类含不溶性颗粒掺杂的体系往往会导致整个体系的不均匀[6],因为连续纤维增强的复合材料具有很高的纤维体积分数,纤维之间的间隙极小,不可避免会影响固化剂的分布和在固化过程中的扩散和分配,造成复合材料内部树脂固化均匀性的差异,进而影响复合材料宏观力学性能。
随着复合材料应用的日益广泛,发展低成本的复合材料制备技术受到关注。应用大丝束纤维、发展大厚度预浸料技术来制备复合材料可以大幅度降低纤维的制造成本、复合材料的铺覆工艺成本,拓展复合材料的应用。大丝束纤维束内纤维的根数可达48000根[7],大厚度的预浸料每平方米质量可达到1600g以上,如此大的厚度势必会影响树脂在纤维层内的渗透,尤其是对于含有大量固化剂和促进剂颗粒的非均相固化体系。因此研究这类固化体系复合材料的内部树脂均匀性非常必要,然而针对这方面的研究还鲜见报道。
纳米力学测试是一种新的微观力学测试方法[8],应用纳米力学测试方法在研究复合材料界面[9]、材料韧性[10]、材料黏弹行为[11-12]等方面取得了不少进展,利用这种方法可以得到材料在微米甚至纳米尺度上的微观力学信息,尤其是纳米硬度等信息[13],在测试微观尺度上的材料力学信息方面具有优势。因此,本工作利用纳米力学的硬度测试方法,结合自带的探针形貌扫描功能,研究具有非均相固化体系树脂的复合材料,包括利用现有的小厚度预浸料(面重960g/m2)和模仿超厚预浸料的真空吸注法制备的复合材料的内部树脂微观力学分布的均匀性。
1 实验材料与方法
1.1 材料及制备
由小厚度预浸料制备的复合材料样品的制备:将S4玻璃纤维的环氧树脂预浸料按对称正交6层铺层,再用真空袋压成型方法和环氧树脂规定的固化工艺固化成型,得到复合材料板,将复合材料板切割成试样,试样表面磨平并抛光。所用的预浸料为S4玻璃纤维和环氧树脂的窄带预浸料,固化条件为125℃/2h;预浸料中的环氧树脂为EDS环氧树脂,EDS环氧树脂为本单位环氧树脂产品,其主要组成为双酚A型环氧树脂和双氰胺固化剂(质量分数为4%);S4 纤维为南京玻璃纤维研究设计院产品,纤维平均直径约10μm,线密度为800g/km。
由树脂真空吸注法制备的复合材料样品的制备:将玻璃纤维紧密缠绕于一金属片上,利用弹簧拉力计控制纤维的牵引力,缠绕完成后纤维末端在金属片卡槽上固定并用胶布进一步固定,避免末端松弛,置于一个铝箔制成的小盒子中,倒入EDS环氧树脂,升温至70℃,再抽真空脱除树脂和纤维孔隙中含有的空气,随后恢复常压使EDS环氧树脂充分浸渍玻璃纤维,最后经过80℃/1h,90℃/1h,125℃/2h的固化工艺固化。冷却至室温后,将复合材料板切割成试样,使纤维方向垂直于切割面,切割面磨平并抛光,备用。所用的玻璃纤维为S6高强玻璃纤维,为南京玻璃纤维研究设计院产品,纤维平均直径约11μm,线密度为480g/km,宽度约为4mm,单束厚度约为85μm。
纳米力学测试试样制备:将复合材料试样切割成上下面平行且高度不超过5mm的小块,并且多数纤维的取向方向垂直于上下面,将其中一面打磨和抛光处理作为测试面,处理工艺为先采用2000目砂纸、400转下细磨10min,再采用金刚石抛光膏抛光45min,最后清洗干燥后得到测试样。
1.2 纳米力学测试
纳米力学测试仪为TI-750Ubi原位纳米力学测试系统,测试纳米硬度时选用Indent模式,施加压力大小为200μN和500μN,接触压力设定为2μN,测试前后分别利用探针扫描观察测试区域的形貌;测试纳米磨损时仍选用Indent模式,只是将接触压力设定为20μN,测试前后分别利用探针扫描观察测试区域的形貌。
1.3 其他测试方法
扫描电镜测试,采用Hitachi扫描电镜,北京市理化测试中心设备,样品表面经喷金处理。
2 结果与分析
2.1 固化体系的颗粒图
双氰胺固化的环氧树脂体系固化温度很高,添加固化促进剂可以显著降低固化温度[14],本工作中的树脂体系采用了一种难溶性的聚脲促进剂。图1给出了EDS环氧树脂的固化剂体系组分的SEM图,分别为固化剂双氰胺(图1(a))和固化促进剂(图1(b))。从图上可以看到,固化剂的颗粒大小不均,但固化剂颗粒多数在1μm以上,部分固化剂颗粒甚至达5μm以上。促进剂颗粒同样大小不均,并存在着明显的聚集,部分促进剂颗粒大小达10μm以上。而对于玻璃纤维复合材料,纤维本身的直径只有10μm左右,纤维之间的间隙甚至小于1μm,根据物理学上的沙漏原理,这在浸渍时必然会造成固化体系的滤过效应,使固化剂和促进剂富集于纤维层外部,固化时仅能通过扩散作用分布到纤维层内部,所以可能会造成不同区域固化程度的不均匀,进而造成内部微观力学的差异。
2.2 由小厚度预浸料制备的复合材料试样的纳米硬度测试
图2给出了光学显微镜下的复合材料截面以及相应的测试位置,整个视野内除了明显地呈现一些黑色的孔隙缺陷,都有较高的平整度和相似性。六层铺层的总厚度为2.90mm,即单层预浸料平均厚度为480μm。考虑到预浸料的制备工艺,单层预浸料由几束玻璃纤维浸渍环氧树脂,并集束而成,因此选取了11个测试区域测试纤维和树脂的纳米硬度作为对比,11个区域分别处于两个铺层的层间或近层间和一个铺层的近中心位置。因集束工艺制备的大厚度预浸料,纤维铺层层间或近层间为固化体系颗粒较大可能富集的区域,而铺层的近中心位置则具有随机性。

图2 光学显微镜下的复合材料截面以及相应的测试区域Fig.2 Optical micrograph of the cross section of the composites (the 11 stars were signed as the selected 11 test regions)
图3为5,6,7,8几个区域的纳米探针扫描图及测试点取点,从图上可以看到各个区域的起伏都不大,抛光面比较平整,树脂区域要略高于纤维区域,高度差均在20nm左右,这表明在抛光处理时树脂的耐磨性要高于玻璃纤维,而各区域高度差均较接近,也表明各区域树脂具有相近的耐磨性。图上的区域5,6为纤维垂直方向的截面,而区域7为两个铺层的层间位置,区域8为纤维横向方向的截面。各区域中,测试点尽量选取离纤维有一定距离的树脂部分,减少纳米探针加载时受到纤维-树脂界面和纤维本体的影响。
图4 给出了区域6中各测试点的载荷-压深曲线,图中的测试点7处于玻璃纤维上,其载荷-压深曲线具有很高的斜率,测试点3可能较靠近纤维,并受到测试系统热漂移定位变化的影响,在压深加大到30nm后,可能压针接触到纤维,导致斜率迅速变大,测试点2远离周围的纤维,受到纤维-树脂界面影响较小,具有相对较小的纳米硬度值;其余测试点具有较好的一致性,在加载后压深达到一个相近的值,并在卸载后部分弹性回复。实验中,每组测试值均去除类似于测试点3和7的结果。

图3 几个测试区域的典型探针扫描图及取点 (a)区域5;(b)区域6;(c)区域7;(d)区域8Fig.3 Typical probe scan images and test points of region (a)region 5;(b)region 6;(c)region 7;(d)region 8
表1汇总了上述11个区域的不同测试点的纳米硬度测试结果的数值分布以及相应的均值和标准差,从表上可以看到,11个区域的纳米硬度均值并没有显著的差异,均主要分布于0.3~0.5GPa内,其各测试点位于0.35~0.4范围内的概率最高。这表明各区域的纳米硬度没有明显区别,这也和图3中的探针扫描图观察到的情况一致。
这11个区域中,区域1,3,5,7,9,11为处于预浸料纤维层边缘的树脂,其所有53个测试点的测试值的纳米硬度为(0.370±0.047)GPa;2,4,6,8,10区域为处于预浸料纤维层中间的树脂,其所有43个测试点的测试值的纳米硬度为(0.383±0.037)GPa。考虑到测试误差影响,两者实际上并没有区别,表明非均相的固化体系并没有对预浸料内部树脂各区域造成显著的力学差异。
2.3 由树脂真空吸注法制备的复合材料试样
图5为由树脂真空吸注法制备的复合材料样品,其中纤维层较厚的样品厚度约2mm,而纤维层较薄的样品厚度约0.5mm,可以看到厚纤维层表面明显有一层白色的物质沉积,表明因渗滤作用在纤维层表面沉积了大量的固化剂、促进剂颗粒,甚至在固化过程中都不能完全溶解,而薄纤维层表面看不到明显沉积,这和纤维层较薄,表面固化剂和促进剂沉积量较少有关。

图4 测试区域6中8个测试点的载荷-压深曲线Fig.4 Load-depth curves of the selected eight test points in region 6
图6为上述厚纤维层复合材料样品垂直于纤维轴向方向的截面的光学显微图以及选择的相应测试区域。从图上可以看到,经过长时间抛光后,其截面上仍然呈现明显的层状分布,这种层状分布是由抛光后的各区域平整度的显著差异造成的。其中区域5为树脂区,具有较高的平整度,区域4为纤维层表面,其平整度低,可能和表面沉积固化剂、促进剂颗粒有关,区域3为浅层纤维区,光学显微镜下可观察到具有较高的平整度,从区域2开始平整度明显变差,观察到出现较多的黑点,而区域1为深层纤维区域,该区域光学反射能力较低,可见大量黑色缺陷点,表明平整度很差,各区域光学视觉的显著差异表明各区域微观特征存在明显差异。而对于利用预浸料制备的复合材料样品,除了一些明显的孔隙缺陷,整个表面都有很高的平整度,表明整体具有较好的均一性。

表1 11个区域的不同测试点的纳米硬度值分布与均值Table 1 Distribution and mean values of nano-hardness of different test points in the 11 regions
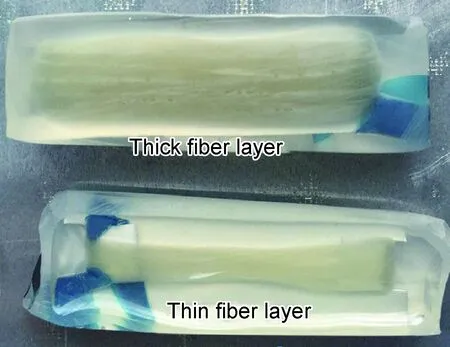
图5 树脂真空吸注法制备的复合材料样品的光学照片Fig.5 Optical image of composites prepared by resin vacuum injection method

图6 光学显微镜下的埋植厚纤维复合材料截面以及相应的测试区域Fig.6 Optical micrograph of the cross section of the composites
图7为上述各区域的纳米压痕探针扫描图,可以看到其表面平整度和光学显微镜观察到的较为一致。对于区域1,几乎看不到树脂,分布着少量残留的树脂和大量的纤维,不易测试树脂的纳米硬度,该区域少数位置可观察到挤出的液滴状或者破碎的树脂,测试时测试这些位置的纳米硬度;对于区域2,可以看到其明显不同于区域1,能观察到固化的树脂,但其高度明显低于纤维,达到了100nm左右,表明该区域树脂的耐磨性很差,在抛光条件下磨损程度明显高于纤维;而对于区域3,树脂区域要高于纤维约20nm,这和图3中的形貌和高度差接近;区域4可观察到较多的白色颗粒,可能为未溶解的固化剂或促进剂的颗粒,实验中测得这些点的纳米硬度在0.1~0.25GPa,显著低于树脂基体,并具有较大的离散性和低的弹性回复,离散性大和颗粒较小、测试误差较大有关,这表明固化体系颗粒在纤维层表面富集,且固化后也不能完全溶解。
表2为各区域树脂的数据统计,图8为不同测试区域中各测试点的典型载荷-压深曲线。对于区域1,树脂的纳米硬度极低,且其载荷-压深曲线在减少载荷时弹性回复很小,表明发生了塑性变形,树脂固化程度很低甚至可能为黏性液体。而区域2的树脂纳米硬度也低于另外3个区域,从其载荷-压深曲线看具有较大的离散,撤去载荷后仍有较大的塑性变形,但形变回复率明显高于区域1。以上表明这两个区域的树脂固化不完全。区域4为纯树脂区,只有部分测试点受到纤维-树脂界面的影响,故其纳米硬度均值要略低于纤维区(区域3)的树脂,同理区域5不受到界面影响,其纳米硬度要更低一点,但仍大于区域2树脂的纳米硬度,但三者的载荷-压深曲线较为接近,撤去载荷后都具有相近的形变回复率。对比以上图表,表明纤维层对颗粒状的固化体系具有明显的滤过效应, 尽管在固化条件下发生了溶解,可使但也仅使纤维表层约0.2mm范围的树脂较好地固化,表层往内0.4mm处则纳米硬度和耐磨损性明显下降,这对大丝束纤维束制备或直接织物和树脂胶膜复合的大厚度预浸料可能仍然会有较大影响,因此对大厚度预浸料的制备工艺具有参考价值,如使用直接织物和树脂胶膜复合的方法,单层或单丝束厚度宜控制在0.4mm以下。

图7 几个测试区域的典型探针扫描图及取点 (a)区域1;(b)区域2;(c)区域3;(d)区域4Fig.7 Typical probe scan images and selected test points of regions (a)region 1;(b)region 2;(c)region 3;(d)region 4
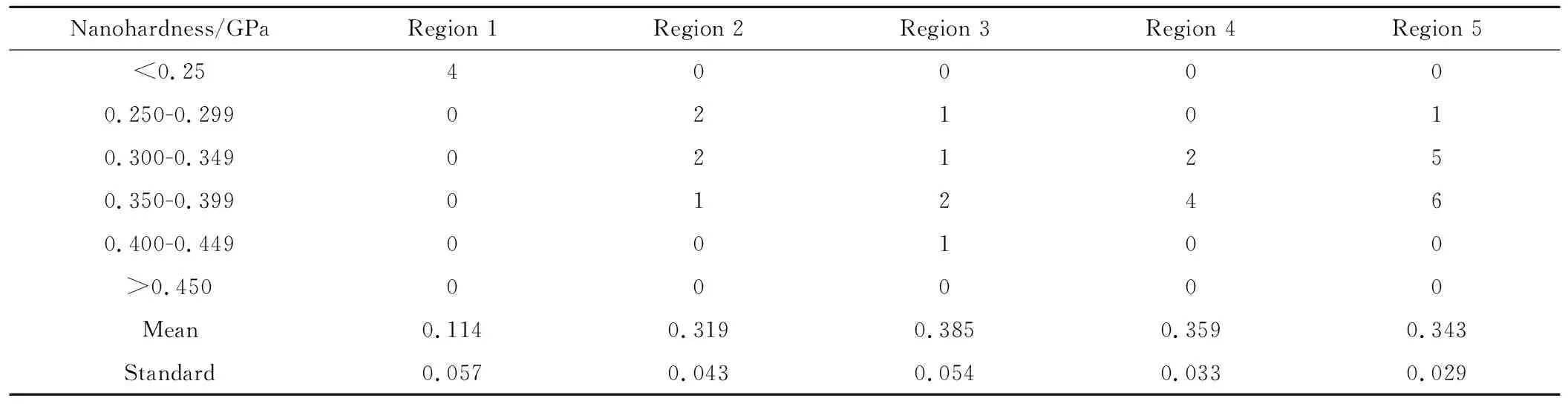
表2 5个区域的不同测试点的纳米硬度值分布与均值Table 2 Distribution and mean values of nano-hardness of different test points in the five regions

图8 不同测试区域中各测试点的典型载荷-压深曲线Fig.8 Typical load-depth curves of the test points on different regions
利用纳米磨损的方法对区域1具有挤出物的部分部位进行磨损测试,见图9,可以看到仅用20μN接触力磨损,就可以把挤出物完全抹去。图9(a)的3根纤维的中心区域树脂和纤维的高度差为75nm,而在图9(b)中,高度差达到了145nm,表明微小的接触力就能使复合材料中的树脂抹除,这同样也表明了该处树脂固化程度极低,力学性能很差。这是因为被滤过的固化剂和促进剂无法通过渗透作用大量扩散到该位置使树脂发生充分固化。

图9 在经过20μN接触力下磨损前后的区域1的探针扫描图 (a)磨损前;(b)磨损后Fig.9 Probe scan images of region 1 before and after wear with a 20μN contact force (a)before wear;(b)after wear
3 结论
(1)对于通过小厚度预浸料得到的复合材料,复合材料内部树脂各区域具有较好的微观力学均匀性,各区域纳米硬度基本相同,其均值均在0.33~0.39GPa之间,抛光后各区域树脂高度均高于纤维约20nm。
(2)对于模仿大丝束大厚度预浸料制备的渗透过程中通过树脂真空吸注法制备的复合材料,固化剂颗粒在纤维层外部富集,距纤维外层约0.2mm内的树脂具有较高的纳米硬度,其值和小厚度预浸料制备的复合材料相当,而再往纤维层内部(≥0.4mm),树脂的纳米硬度下降且易磨损,纤维层较深处则树脂固化不完全,纳米硬度很低,表明复合材料内部树脂各区域具有较差的微观力学均匀性。
猜你喜欢
河北地质(2022年2期)2022-08-22
水泥工程(2022年2期)2022-08-22
建材发展导向(2022年12期)2022-08-19
军民两用技术与产品(2022年1期)2022-06-01
北京航空航天大学学报(2021年9期)2021-11-02
上海大学学报(自然科学版)(2020年4期)2020-05-24
环境科技(2016年1期)2016-11-08
中国塑料(2016年3期)2016-06-15
浙江大学学报(工学版)(2016年9期)2016-06-05
工程建设与设计(2016年8期)2016-03-11
