
超高效液相色谱串联质谱法同时测定饮料中γ-羟基丁酸及其前体物质
2018-10-17 11:07龚蕾韩智刘杰朱晓玲王会霞彭青枝
食品与发酵工业 2018年9期
龚蕾,韩智,刘杰,朱晓玲,王会霞,彭青枝
(湖北省食品质量安全监督检验研究院,湖北 武汉,430075)
γ-羟基丁酸(GHB)具有强烈的镇静作用和健忘效果,并且无色无味,其钠盐稳定存在,很容易加入到饮料中而不被发觉;同时,GHB也能刺激人体分泌荷尔蒙素,增加快感,因此GHB往往又与性犯罪联系在一起,在娱乐场所被滥用,带来了严重的社会问题[1]。欧美很多国家已经将GHB列为一类药品管制,我国于2001年5月将GHB列为二类精神药物进行管理,2007年我国将其列入一类精神药物进行管制[2]。但是由于原料易得和合成简单,其违法使用情况依然存在,此现象引起了人们的广泛重视。γ-丁内酯(GBL)作为一种食品添加剂可用于食品合成香料领域,1,4-丁二醇(1,4-BD)可作为食品接触材料添加剂运用于包装材料中。然而,GBL在酶催化作用下或提高pH情况下,可以很快转化为GHB;1,4-BD在人体中也很快代谢为GHB,3种物质的结构式见图1。
目前并没有同时检测食品中GHB、1,4-BD及GBL的国家标准,只有一个公共安全行业标准GA/T 1074—2013《生物样品中γ-羟基丁酸的气相色谱-质谱和液相色谱-串联质谱检验方法》[3],但该标准的检测对象为生物样品(尿液、血液、组织和毛发),检测物质仅是GHB。据文献报道,GHB的分析方法主要有气质联用法(GC-MS)[4-6]、液质联用法(LC-MS)[4, 7-10]和毛细管电泳法(CE)[11]等。气相色谱法(GC)应用较少,主要是由于GHB分子量小,且含有一个羟基和一个羧基两个强极性基团,热稳定性不好。与GC相比,GC-MS应用较为广泛,但是一般也需要衍生化处理或将GHB转化为GBL,因此也只能检测一种物质。WOOD等[12]以C18为固定相,以甲醇-0.1%(体积分数)甲酸水溶液为流动相,对尿液中的GHB、GBL、1,4-BD进行了分析,检出限为1 mg/L。张浩杰[13]将饮料用滤膜过滤后,用T-3色谱柱分离,以甲醇-水为流动相梯度洗脱,串联质谱MRM模式检测,5 min内实现有效分离。施妍等[14]利用HPLC-MS/MS测定尿液中的GHB、1,4-BD和GBL,检出限分别为0.1、0.1和2μg/mL,准确度为87.6%~98.1%。

图1 GHB(a), GBL(b)和1,4-BD(c)的化学结构Fig.1 Chemical structures of GHB, GBL and 1,4-BD
目前,针对生物样品例如尿液血液中的GHB含量检测已经有相关文献报道[5-10]。然而,由于GBL和1,4-BD在人体中都可以很快代谢成GHB,并且GHB自身在体内也会很快代谢,导致生物样品中GHB及其相关物质的检测具有较强的时效性,从而给实际案件的鉴定和确证工作带来了很大的不确定性。而且,GHB内源性的天然物质,国际毒理学会建议人体尿样中GHB的临界值(cut-off值)为10 μg/mL,这给实际尿样阳性结果的判断带来一定困难。GHB,GBL和1,4-BD经常被犯罪分子故意添加到饮料中进行犯罪活动,而三者在饮料中不会发生代谢,稳定性较高,因此有必要建立饮料中GHB,GBL和1,4-BD的含量检测方法,为GHB及其前体物质的合法性提供数据。本文建立了饮料中GHB,GBL和1,4-BD同时检测的UPLC-MS/MS分析方法,此方法快速准确,是一种方便且可靠的检测手段。
1 材料与方法
1.1 仪器与试剂
Water Xevo-TQD三重四级杆质谱仪,美国Waters公司;电子天平,梅特勒-托利多仪器(上海)有限公司;P型移液器,德国Brand公司;离心机,美国Beckman Coulter公司;0.22 μm滤膜,天津津腾实验设备有限公司。
γ-羟基丁酸钠(纯度≥98.0%),美国MedChemExpress公司;γ-丁内酯(纯度≥99.9%),北京坛墨质检科技有限公司;1,4-丁二醇(纯度≥99.6%),德国Dr. Ehrenstorfer GmbH公司;乙酸铵、甲酸(色谱纯),美国Fisher公司;乙腈、甲醇(色谱纯),德国Merck KgaA公司;饮料样品购置于网络平台或市售,包括固体饮料(按照产品推荐的冲调倍数稀释)。
GHB、1,4-BD、GBL单标溶液:分别称取100 mg标准品于100 mL容量瓶中,用超纯水稀释并定容到刻度,配制成1 mg/mL的单标溶液,备用。
GHB、1,4-BD、GBL基质标准溶液:分别称取一定体积GHB、1,4-BD、GBL单标溶液于10 mL容量瓶中,用稀释了25倍的空白样品提取液定容到刻度,配制成0.5、1.0、2.5、5.0、10.0μg/mL的基质标准溶液,备用。
1.2 UPLC-MS/MS条件
1.2.1 UPLC条件
Thermo Gold C18(150 mm×2.1 mm,3.0 μm)色谱柱;流动相:A为2 mmol/L乙酸铵水溶液,B为乙腈,梯度洗脱程序见表1;流速0.2 mL/min;柱温30 ℃;进样量5 μL。
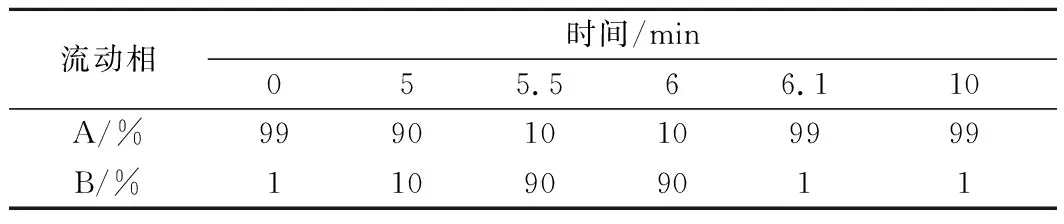
表1 梯度洗脱程序Table 1 The gradient elution program
1.2.2 质谱条件
离子源:电喷雾离子源(ESI);扫描方式:正负离子模式同时扫描;毛细管电压:2 000 V;源温度:120 ℃;脱溶剂温度:300 ℃;脱溶剂气流速:800 L/h;锥孔气流量:30 L/h;采集模式:多反应监测(MRM)。
1.3 样品前处理
1.3.1 不含蛋白饮料
称取1 g样品(精确至0.01 g)于25 mL容量瓶中,加水稀释至刻度,混匀,经微孔滤膜过滤,取续滤液,根据实际浓度适当稀释至线性范围内,供UPLC-MS/MS测定。
1.3.2 含蛋白饮料
称取1 g样品(精确至0.01 g)于25 mL容量瓶中,加入2 mL甲醇,摇匀,以沉淀蛋白质,再加水定容至刻度,混匀,转移至离心管中于4 500 r/min下离心15 min,取上清液,经微孔滤膜过滤,取续滤液,根据实际浓度适当稀释至线性范围内,供UPLC-MS/MS测定。
2 结果与讨论
2.1 质谱条件的优化
采用蠕动泵分别将质量浓度为10 μg/mL的GHB、1,4-BD、GBL单标溶液注入离子源,以便于对质谱分析条件进行优化。在全扫描方式下分别对3种化合物进行一级质谱分析,发现1,4-BD和GBL在正离子模式下响应较高,GHB在正离子和负离子模式下均有响应,但据后续试验发现,同时测定3种化合物需要采用正负离子同时扫描。本实验GHB采用负离子扫描模式,1,4-BD和GBL采用正离子扫描模式,并在各自对应的模式下得到各自的分子离子峰,通过优化锥孔电压使各分子离子峰达到最大响应值;再分别对分子离子进行二级质谱分析(子离子扫描),得到相应的碎片离子,并通过优化碰撞能量使碎片离子达到最大响应值。得到最佳质谱条件见表2。
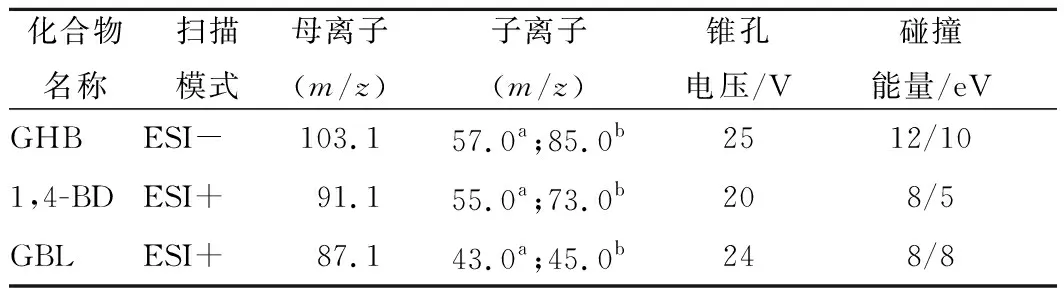
表2 三种化合物的定性离子、定量离子、锥孔电压 和碰撞能量Table 2 Qualitative ion, quantitative ion, cone voltage and collision energy of three compounds
注:a-定量离子;b-定性离子
2.2 源参数优化
图1展示了GHB和GBL的化学结构式,GBL是由GHB脱去1分子水缩合而成的环状化合物。有国外文献报道了GHB和GBL在源内的相互转化[7, 9],即“源内裂解”,而已有的国内文献则并未提及。源内裂解主要是指由于离子化过程较为剧烈,使得生成的离子具有足够的内能发生进一步裂解,如失H2O、失糖苷等,生成的基团(子离子)与某种物质的结构一样而被作为“母离子”继续被裂解。源内裂解的过程与四级杆MS/MS的裂解过程十分相似,后者主要是使用一个相对较低的碰撞能量来产生碎片,其观察到的碎片离子依赖于碰撞能量的大小,而源内裂解的过程主要依赖于源区的接口电压的大小,一般而言,源内裂解只能减少而不能消除。在本试验中也观察到了源内裂解现象,因此,降低源内裂解程度是本试验所关注的重点。研究表明,降低源温度及喷雾电压能有效减少源内裂解程度,但同时会降低离子丰度。本试验综合考虑得出最佳条件为:源温度120 ℃,喷雾电压2 000 V,脱溶剂温度300 ℃,脱溶剂气流速为800 L/h。
GHB在ESI+模式下失水产生87.0的碎片离子,此碎片离子的质量数与GBL在ESI+模式下的一级母离子质量数一致;GBL则容易加水产生105.0的离子,此离子与GHB在ESI+模式下的一级母离子质量数一致,若同时采用ESI+模式扫描,则不能从色谱图区分2种化合物,见图2-e与图2-f。因此,GHB采取ESI-模式扫描,GBL与1,4-BD采用ESI+模式扫描,能从色谱图上区分开来。由2-a为GHB的单标标准溶液进样后得到的总离子流图(TIC),图2-b为GBL的单标标准溶液进样后得到的总离子流图,从图2-a中可以看出GHB单标标准溶液色谱图中,在相同保留时间(3.13 min)的地方也出现了GBL的峰,图2-b中GBL的单标标准溶液的色谱图中显示GBL的出峰时间为3.70 min,此结果显示GHB易发生源内裂解,与JOHANSEN[7](图2-c)和SØRENSEN[9](图2-d)的研究结果一致。因此,建议采取合适的洗脱梯度将GBL与GHB完全分离。


a-GHB单标TIC图;b-GBL单标TIC图;c-JOHANSEN研究结果;d-SØRENSEN研究结果;e-ESI+模式下GHB的TIC图;f-ESI+模式下的GBL的TIC图图2 源内裂解相关色谱图Fig.2 Some chromatograms of in-source collision-induced dissociation
2.3 三种物质的定量分析方法考察
2.3.1 色谱柱的选择
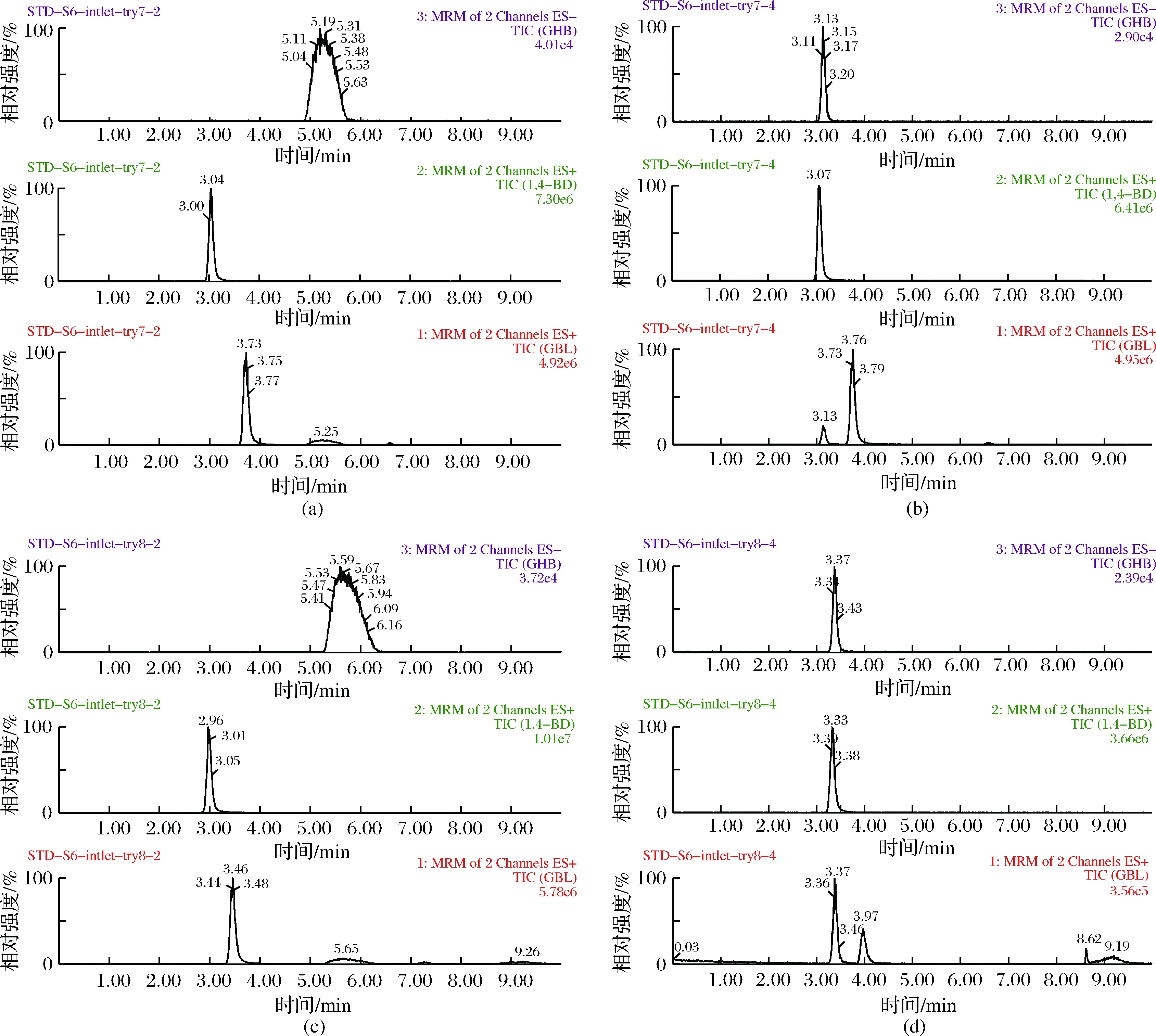
本研究中的3种目标化合物中,GHB为极性化合物,1,4-BD为弱极性化合物,GBL为非极性化合物,因此,在选择色谱柱时,应兼顾保留极性化合物同时对中等极性和非极性化合物也应有所保留。本研究比较考察Waters Acquity UPLC BEH C18(2.1 mm×100 mm, 1.7 μm)、Waters Atlantis T3(2.1 mm×50 mm, 1.7 μm)、Thermo Gold C18(150 mm×2.1 mm, 3.0 μm)三种实验室常用的色谱柱,结果表明,GHB、1,4-BD在前两种色谱柱上保留较差,而Thermo Gold C18(150 mm×2.1 mm, 3.0 μm)色谱柱对GHB、1,4-BD有保留,峰型较好(图3)。因此,本研究初步选择 Thermo Gold C18(150 mm×2.1 mm, 3.0 μm)色谱柱开展后续实验。
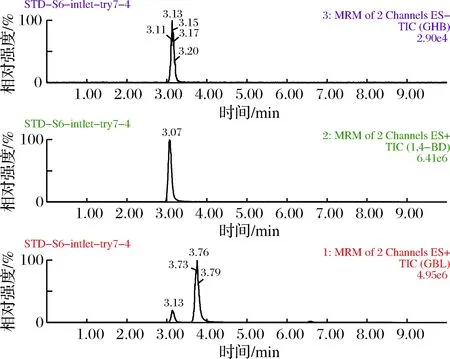
图3 GHB、1,4-BD和GBL总离子流图Fig.3 Total lon chromatograms of GHB, 1,4-BD and GBL
2.3.2 流动相的选择
液相使用的流动相为乙腈和2 mmol/L乙酸铵溶液。前期比较考察了乙腈-水(图4-a)、乙腈-2 mmol/L乙酸铵溶液(图4-b)、甲醇-水(图4-c)和甲醇-2 mmol/L乙酸铵溶液(图4-d)4个流动相体系,实验结果表明,当采用乙腈时,所得到的色谱峰峰型较尖锐,因此选择乙腈作为流动相体系之一;当采用水作为流动相时,GHB出峰时间延后,但其色谱峰较宽,峰形较差,将水相中加入甲酸或乙酸铵后,其色谱峰峰形明显改善。另外,鉴于本实验质谱部分所采用的扫描模式为正负离子同时扫描,所以选择低浓度的乙酸铵溶液作为流动相可有效避免甲酸对负离子扫描的抑制作用,同时避免高浓度的盐可能会造成的离子竞争抑制。因此,最终选择以乙腈-2 mmol/L乙酸铵溶液作为流动相。
2.3.3 基质效应
UPLC-MS/MS是水溶液中药物残留的首选检测方式之一,但是这种检测方式的一个主要缺点是具有基质效应(matrixeffects,ME)。一般来说,当ME在80%~120%时,表明基质效应在可接受范围内,在实际检测中可以忽略基质效应。本研究比较了样品直接过膜上机和将样品进行一定倍数的稀释后再上机检测两种处理方法,并比较了不同稀释倍数对检测结果的影响。
参照张浩杰等[13]对饮料的处理方式以及后期优化前处理条件进行处理分析,表3为样品的加标回收率。可以看出,直接过膜上机对GHB和1,4-BD的回收率较低,不满足实验要求,同时也表明,饮料基质对GHB和1,4-BD的检测存在较强的基质干扰。
a-乙腈-水;b-乙腈-2 mmol/L乙酸铵;c-甲醇-水;d-甲醇-2 mmol/L乙酸铵图4 不同流动相的影响Fig.4 The effects of different flow phases

表3 GHB、1,4-BD、GBL回收率(直接过膜上机) 单位:%
基质效应的消除可采取配制基质标准曲线或对样品进行其他前处理净化等方式解决。鉴于GHB、1,4-BD和GBL的极性差异较大,分别为极性、弱极性、非极性化合物,因此,较难选择合适的固相萃取小柱进行净化处理。同时,γ-丁内酯在前处理过程中可随水蒸气挥发,因此,对于碳酸饮料和含酒精的饮料也不适合采用加热除二氧化碳或除酒精后复溶的处理过程。考虑到饮料中若非法添加GHB、1,4-BD、GBL,根据不法分子的添加动机分析,其添加量不可能很低(因为GHB在人体中本身就内源性存在,添加量太少,对人体不起作用),所以本研究最终选择直接稀释的方法进行样品处理以消除基质效应。
本实验将样品分别稀释5倍、10倍、25倍及50倍进行加标回收,结果表明(图5),随着稀释倍数增加,GHB的基质效应显著降低,但依然不高于50%;当稀释倍数达到25时,1,4-BD和GBL回收率均超过90%,基质效应可基本忽略。因此,在实际样品检测时,建议考虑按照1.1配制基质标准曲线,以消除不同基质对质谱中GHB响应信号的影响。考虑到方法检出限、仪器响应等情况,最终将稀释倍数定为25倍。

图5 不同稀释倍数下GHB(a)、1,4-BD(b)、GBL(c)加标回收率%Fig.5 The recovery of GHB(a), 1, 4-BD(b) and GBL(c) of different dilution multiple
2.3.4 线性关系和检出限
配制一系列质量浓度的标准溶液在2.2的仪器分析条件下进行测定,得出3种物质在0.5~10.0 μg/mL呈现出良好的线性关系,相关系数r均大于0.99,检出限(RSN=3)均为0.5 μg/mL。3种化合物的标准曲线、线性范围和相关系数见表4。

表4 GHB、1,4-BD、GBL的线性范围、校准曲线 方程和相关系数Table 4 Linear range, calibration curve equation and correlation coefficient of GHB, 1, 4-BD and GBL
2.3.5 精密度、重复性和稳定性
准确吸取标准品溶液,按2.2的仪器条件进样,连续进样6次,测定各自的峰面积,以峰面积为对象,计算相对标准偏差(RSD)。计算得标准品峰面积RSD小于0.10%,保留时间RSD小于1.0%,表明该方法精密度良好。
平行制备同一浓度混合标准品6份,按2.2的仪器条件进样,以标准品的峰面积和保留时间为对象,得出RSD值均小于1.5%,表明该测定方法的重复性良好。
准确吸取质量浓度为10 μg/mL的标准品溶液,在1.2的仪器条件下,分别在0、1、2、4、8、12、24、36、48 h进样,以标准品的峰面积和保留时间为对象,得出RSD<2.0%,表明标准品溶液在48 h内稳定,仪器稳定性良好。
2.3.6 回收率
称取同一样品各3份,分别对样品进行3个水平的加标回收率实验,每个实验均6次重复,以不加混合标准品溶液的样品作参照,并按照2.3方法制备供试品溶液,按2.2的仪器条件进样,根据测定的含量,计算其相应的回收率,结果见表5。不含蛋白饮料GHB的回收率为81.1%~87.7%,RSD为3.8%~5.7%(n=6),1,4-BD的回收率为98.5%~99.6%,RSD为4.4%~5.3%(n=6),GBL的回收率为107.5%~113.6%,RSD为3.9%~5.1%(n=6);含蛋白质饮料GHB的回收率为82.6%~82.8%,RSD为3.3%~5.2%(n=6),1,4-BD的回收率为98.7%~99.8%,RSD为3.7%~4.4%(n=6),GBL的回收率为110.5%~116.2%,RSD为3.5%~4.6%(n=6)。结果表明上述实验方法准确度较高,能应用于饮料中GHB、1,4-BD和GBL的检测分析。

表5 GHB、1,4-BD、GBL的回收率(n=6)Table 5 The recovery of GHB, 1, 4-BD and GBL(n=6)
2.4 实际样品检测
采用所建立的方法对市售和网上购买的样品(共40批次)进行了检测,结果如表6。
3 结论
本研究建立了饮料中GHB及其前体物质1,4-BD和GBL的HPLC-MS/MS方法,其检出限为0.5 μg/mL,该方法灵敏度高、简便快速,可靠性高。应用此方法,本研究检测了40份样品中GHB、GBL和1,4-BD的含量水平,检验结果均为未检出。本研究所建立的分析方法及研究结果能为实际检测提供技术支持和数据支撑。

表6 市售和网购各类饮料中GHB、1,4-BD和GBL的测试结果Table 6 The result of GHB, 1, 4-BD and GBL in actual beverage samples
注:ND代表未检出。
猜你喜欢
中老年保健(2022年3期)2022-11-21
娃娃乐园·综合智能(2022年7期)2022-07-16
煤化工(2022年3期)2022-07-08
色谱(2021年7期)2021-06-07
食品安全导刊(2020年21期)2020-09-07
农家科技中旬版(2019年9期)2019-10-08
山西农业科学(2019年6期)2019-06-19
小学生学习指导(低年级)(2018年5期)2018-04-24
小天使·一年级语数英综合(2017年1期)2017-02-16
山东工业技术(2016年13期)2016-06-29
