
基于高通量测序和可培养方法的勐海发酵普洱茶细菌多样性分析
2018-10-17 11:16:18张欣姚粟白飞荣田海霞赵婷马跃刘海新李颂郝彬秀王春玲
食品与发酵工业 2018年9期
张欣,姚粟*,白飞荣,田海霞,赵婷,马跃,刘海新,李颂,郝彬秀,王春玲
1 (中国食品发酵工业研究院有限公司,中国工业微生物菌种保藏管理中心,北京,100015) 2 (中国茶叶有限公司,北京,102209)
勐海是云南地理标志产品普洱茶的重要发祥地和产区[1-2]。发酵普洱茶是采用“渥堆发酵技术”,以地理标志保护范围内的云南大叶种为原料,经杀青、揉捻、晒干制成晒青毛茶,再经人工洒水,渥堆微生物发酵之后而成的后发酵茶类,具有独特的风味和显著的保健功能,深受大众喜爱[3-5]。
微生物是普洱茶后发酵的核心,对普洱茶的品质产生至关重要的影响。不同原料、不同环境和不同的工艺造就了微生物的多样性变化,通过微生物的代谢,产生多种生化产物,形成了普洱茶多样化的风味[6]。目前,关于普洱茶发酵微生物的研究多集中在真菌,如黑曲霉、青霉、木霉等[7-10],对普洱茶发酵中细菌的研究较少。姚静等人从普洱茶渥堆发酵过程中分离到芽胞杆菌属、克雷伯氏菌属、短杆菌属、假单胞菌属等7个属的细菌[11]。王辉等人发现大理南涧凤凰沱茶厂普洱茶堆中存在欧文氏菌属、无色杆菌属、芽胞杆菌属、类芽胞杆菌属、鞘氨醇杆菌属、短杆菌属、葡萄球菌属等优势细菌[12]。
对普洱茶微生物种类的研究主要依靠传统的平板分离、纯化等可培养的方法。但传统方法存在数据不完整,不能全面揭示发酵过程中微生物的群落组成。随着分子生物学的发展,PCR-DGGE、克隆文库等非培养技术[13-15]开始用于普洱茶发酵微生物群落研究。Illumina高通量测序是近年来新兴起来的基于宏基因组的深度测序技术,能直接反映微生物群落构成和多样性,为环境、传统发酵食品的微生物解析提供了新的工具,具有通量高、全面、准确的优势[16-17]。
本文基于高通量测序的非培养技术,并结合传统可培养方法对勐海普洱茶发酵各阶段细菌种类、群落多样性进行了研究,旨在揭示普洱茶发酵过程中细菌群落组成和变化规律,为普洱茶生产工艺的优化提供理论依据。
1 材料与方法
1.1 材料
1.1.1 茶叶样品
普洱茶样品采集自云南勐海百中堂茶庄(经纬度E 100°27′13″,N 21°58′57″;海拔1 136 m;环境温度19~30 ℃,湿度45%~97%)同一发酵茶堆的9个发酵节点,具体信息见表1。取样方法:利用取样钎在发酵堆5个位点(四周及中央)的表面和内部(表面下约20 cm处)各取约200 g发酵茶样品,置于取样袋中充分混匀,冷链运至实验室后,将样品分成两等份,1份4 ℃保存,用于可培养方法分离菌种,1份-80 ℃保存,用于基因组DNA提取和高通量测序。

表1 样品编号及描述Table 1 Numbers and description of samples
1.1.2 试剂与仪器
试剂:胰酪胨大豆琼脂(TSA)培养基,美国BD公司;磁珠基因组提取试剂盒,美国Omega Bio-Tek公司;细菌基因组DNA提取试剂盒,天根生化科技(北京)有限公司;PCR MasterMix、DL 2000 Marker,北京全式金生物技术有限公司;PCR引物由生工生物工程(上海)股份有限公司合成。
仪器:高速冷冻离心机(5424R),德国Eppendorf公司;PCR仪(TRIO48),德国Biometra公司;电泳仪(EC250-90),美国BIO-RAD公司;恒温培养箱(GHP-9160),上海一恒科学仪器有限公司;测序由北京诺赛基因组研究中心有限公司完成;Illumina Miseq高通量测序由深圳华大基因科技有限公司完成。
1.2 方法
1.2.1 茶叶样品可培养细菌的分离与鉴定
1.2.1.1 茶叶样品可培养细菌的分离
将茶样按四分法缩分后准确称取25 g,置于225 mL灭菌生理盐水中,150 r/min振荡1 h,制成10倍稀释液,取1 mL 稀释液于9 mL灭菌生理盐水的试管中,依次稀释后制成 10-2、10-3、10-4、10-5、10-6浓度梯度稀释液,选取10-3、10-4、10-5、10-6四个稀释度,用移液器分别吸取0.l mL稀释液涂布于TSA平板培养基上,每个稀释度3个平行,于(28±1)℃培养箱内倒置培养48 h。根据菌落大小、形态和颜色等表型特征挑选优势菌落进行平板划线分纯,获得纯菌种,并于-80 ℃甘油管冷冻保藏备份。
1.2.1.2 茶叶样品可培养细菌的分子鉴定
利用细菌基因组DNA提取试剂盒提取优势菌株DNA,以通用引物:27F(5′-AGAGTTTGATCCTGGCTCAG-3′)和1492R (5′-GGTTACCTTGTTACGACTT-3′)[18]扩增菌株16S rDNA序列,再经EzBioCloud数据库鉴定[19]。反应条件及序列数据比对分析参照ZHANG等人的方法[20]。
1.2.2 茶叶样品的Illumina高通量测序分析
1.2.2.1 茶叶样品DNA提取及高通量测序
液氮研磨结合磁珠法DNA提取试剂盒提取不同工艺阶段茶叶样品DNA,每个样品重复提取3次并混匀。用微量核酸蛋白分析仪检测DNA浓度及纯度。采用细菌16S rDNA V4区引物进行PCR扩增[21]。利用Illumina高通量测序仪对茶样DNA PCR扩增产物进行测序分析。
1.2.2.2 高通量测序数据分析
利用Illumina MiSeq 2×250 bp paired-end 测序后,下机数据经过滤,去除低质量的reads,用高质量的Clean data进行后续分析[22]。通过软件FLASH,将reads通过reads之间的Overlap关系拼接成Tags[23]。通过软件USEARCH在97%的相似度下将Tags聚成操作分类单元(Operational Taxonomic Unit,OTU)[24]。通过R(v3.1.1)语言中的VennDiagram包绘制Venn图(不考虑OTU丰度,只考虑OTU有无),给出样品间共有OTU个数。通过软件RDP classifer(v2.2)将OTU代表序列在数据库(Greengene V201305; RDP Release9 201203)中比对,进行OTU物种注释[25]。通过软件QIIME(v1.80)对样品进行组成差异分析并计算样品间距离,以判断各样品物种组成的差异性[26]。基于OTU和物种注释结果进行样品物种多样性以及样品间物种差异分析。
2 结果与分析
2.1 茶叶样品可培养优势细菌的分离与鉴定
采用平板稀释法,选择合适稀释度涂布的平板,根据菌落形态、大小、颜色等表型特征,挑取不同单菌落,再经平板划线分纯,共获得76株细菌菌株。经16S rDNA序列测定及比对分析,获得优势菌株的种属名称。部分优势菌株的菌落形态及显微形态见图1,不同茶样获得优势菌株的16S rDNA鉴定结果见表2,序列登录号MG557666~MG557688。
9个样品分离到细菌18个属,30个种,共计76株。分离到的优势菌主要分布在微杆菌属(Microbacteriumsp.)、葡萄球菌属(Staphylococcussp.)、小短杆菌属(Brachybacteriumsp.)、考克氏菌属(Kocuriasp.)、泛菌属(Pantoeasp.)、芽胞杆菌属(Bacillussp.)等。发酵起始阶段晒青毛茶原料(G2)溶出大量内生菌,分离到的细菌种类较多。随着发酵开始,环境微生物和暴露的内生细菌在外界温湿度条件下,选择性增殖,此消彼长。渥堆发酵开始后至发酵7 d(G3、G4),主要优势菌逐渐集中在泛菌属(Pantoeasp.)、Pluralibactergergoviae和假单胞菌属(Pseudomonassp.)。发酵15 d(G5)优势菌群转变为微杆菌属(Microbacteriumsp.)和葡萄球菌属(Staphylococcussp.),并稳定维持到发酵完成(G10),提示这2种优势细菌对于普洱茶后发酵的作用需要重点研究。另外,16S rDNA鉴定结果表明,分离到的微杆菌属(Microbacteriumsp.)优势菌株均为新种,小短杆菌属(Brachybacteriumsp.)、考克氏菌属(Kocuriasp.)和泛菌属(Pantoeasp.)也分布有新种,这些发酵普洱茶中发现的潜在新种资源在国际上尚属首次,对其功能和分类研究具有重要意义。

图1 部分优势菌株的菌落形态(a)及显微形态(b)Fig.1 Colony morphology and microscopic morphology of some dominant strains
表2 不同茶样分离的优势菌种Table 2 The dominant species isolated from different tea samples

样品编号菌落总数/(CFU·g-1)优势菌种G21.5×105Kocuria palustris、Kocuria koreensis、Pantoea sp.、Oceanobacillus iheyensis、Brevibacterium sp.、Brachybacteri-um sp.、Bacillus sp.、Brevundimonas sp.、Paracoccus yeei、Pseudomonas sp.G36.3×108Pantoea sp.、Pluralibacter gergoviae、Pseudomonas sp.、Brachybacterium sp.、Klebsiella sp. G45.0×109Pantoea sp.、Pluralibacter gergoviae、Pseudomonas sp.G56.5×109Listeria grayi、Kocuria koreensis、Microbacterium sp.、Staphylococcus sp.、Alcaligenes sp.、Serratia sp.、Mi-crococcus sp. G62.3×109Microbacterium sp.、Staphylococcus sp.、Brachybacterium sp.G7/Microbacterium sp.、Staphylococcus sp.、Brachybacterium sp.G8/Microbacterium sp.、Staphylococcus sp.、Brachybacterium sp.G9/Microbacterium sp.、Staphylococcus sp.、Paracoccus communisG103.3×109Kocuria koreensis、Microbacterium sp.、Staphylococcus sp.
2.2 茶叶样品的Illumina高通量测序分析
2.2.1 OTU统计及物种注释
利用Illumina Miseq高通量测序技术对9个发酵时间节点的茶样DNA PCR产物进行测序,测序数据经软件拼接、过滤等处理后,9个样品共获得246 297条序列。在97%相似度下将其聚类用于物种分类的OTU(Operational Taxonomic Units), 9个样品共产生212个OTU。OTU的venn图(图2)直观展示了样品间OTU的重叠情况。通过分析,G2、G4、G5、G6和G7共有的OTU为22;G6、G7、G8、G9和G10共有的OTU为77;G7、G8、G9和G10共有的OTU为88;G8、G9和G10共有的OTU为89,分别占这3个样品细菌OTU的62%、66%和75%;G9和G10共有的OTU为95,占这2个样品细菌OTU的70%和80%。说明随着发酵的进行,微生物群落构成趋于稳定。
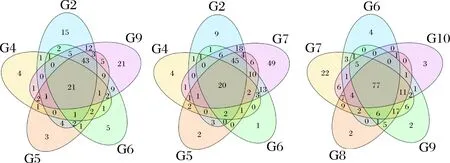
图2 OTU venn分析Fig.2 OTU venn analysis
将OTU代表序列在数据库中比对,进行物种注释。9个样品的细菌主要分布在61个属,28个科,4个门(放线菌门(Actinobacteria)、拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)和变形菌门(Proteobacteria))。
2.2.2 发酵不同阶段的菌落构成及丰度
根据OTU在发酵不同阶段的每个样品的丰度信息,计算出每个OTU在各个样品中的相对丰度(即各种细菌物种所占样品细菌总数的比例)。结果如图3和表3所示。
其中属水平丰度>5%的有12个属:乳杆菌属(Lactobacillussp.)、葡萄球菌属(Staphylococcussp.)、考克氏菌属(Kocuriasp.)、土地杆菌属(Pedobactersp.)、草螺菌属(Herbaspirillumsp.)、短杆菌属(Brevibacteriumsp.)、欧文氏菌属(Erwiniasp.)、泛菌属(Pantoeasp.)、假单胞菌属(Pseudomonassp.)、芽胞杆菌属(Bacillussp.)、肠杆菌属(Enterobactersp.)和小短杆菌属(Brachybacteriumsp.)。
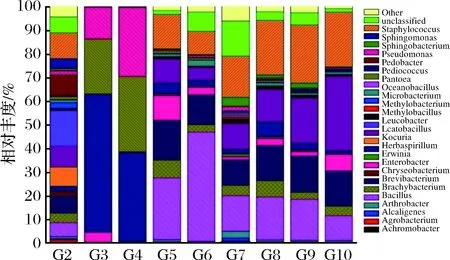
图3 样品属水平的物种profiling堆积柱状图Fig.3 Profiling stack column of the sample at the levelof the genus
表3 发酵不同阶段各样品优势菌属及丰度Table 3 Dominant genera and abundance of samples in different stages of fermentation

样品编号OTU数目优势菌种及丰度G2111Lactobacillus sp.(15%); Staphylococcus sp.(11%); Kocuria sp.(9%); Pedobacter sp.(9%); Herbaspirillum sp.(8%); Brevibacterium sp.(7%); Brachybacterium sp. (4%)G350Erwinia sp.(58%); Pantoea sp. (23%); Pseudomonas sp. (13%)G439Erwinia sp.(38%); Pantoea sp. (32%); Pseudomonas sp. (29%)G599Bacillus sp. (26%); Brevibacterium sp. (17%); Staphylococcus sp. (15%); Enterobacter sp. (11%); Kocuria sp. (10%); Brachybacterium sp. (7%); Microbacterium sp. (1%)G6105Bacillus sp.(46%); Brevibacterium sp. (12%); Staphylococcus sp. (10%); Kocuria sp. (6%); Brachybacterium sp. (3%); Microbacterium sp. (2%)G7179Staphylococcus sp.(17%); Bacillus sp.(15%); Kocuria sp. (11%); Brevibacterium sp. (11%); Brachybacterium sp. (4%)G8143Staphylococcus sp.(23%); Bacillus sp.(18%);Brevibacterium sp. (15%); Kocuria sp. (14%);Brachybacterium sp. (7%)G9135Staphylococcus sp.(25%); Kocuria sp. (19%); Brevibacterium sp. (15%); Bacillus sp.(17%); Brachybacterium sp. (3%)G10119Kocuria sp. (31%); Staphylococcus sp.(23%); Brevibacterium sp. (15%); Bacillus sp.(10%); Brachybacterium sp. (4%)
晒青毛茶原料样品(G2)菌落构成较为丰富,包括乳杆菌属(Lactobacillussp.)、葡萄球菌属(Staphylococcussp.)、考克氏菌属(Kocuriasp.)、土地杆菌属(Pedobactersp.)、草螺菌属(Herbaspirillumsp.)及短杆菌属(Brevibacteriumsp.)。渥堆发酵开始后(G3、G4),革兰氏阴性的欧文氏菌、泛菌属和假单胞菌大量繁殖,占90%以上。渥堆发酵15 d至发酵完成(G5至G10),葡萄球菌、芽胞杆菌、考克氏菌、短杆菌成为整个发酵中、后期的优势菌群,优势菌种此消彼长。而短杆菌和小短杆菌在整个过程中相对维持稳定(分别约占菌群15%和5%)。发酵中期(发酵15 d(G5)、发酵21 d(G6)),芽胞杆菌最为优势。发酵后期(发酵29~41 d(G7、G8、G9)),芽胞杆菌优势度逐渐下降,葡萄球菌跃升为最优势菌,考克氏菌逐渐增加。发酵完成(G10),葡萄球菌维持稳定,芽胞杆菌优势度继续下降,考克氏菌进一步增殖,成为最优势菌种。
2.2.3 样品间物种组成差异分析
根据各样品差异性的统计结果,对样品进行Beta多样性(Beta diversity)分析,比较样品间在物种多样性方面存在的差异大小,见图4。采用Bray-Curtis距离作为衡量群落之间差异性的指标,Bray-Curtis距离值在0~1之间,值越大表示样品间的差异越大。 由图4可知,发酵前期样品(G3、G4)差异最小,发酵中期样品(G5、G6)组成相似,发酵后期样品(G7、G8、G9和G10)聚为一支,组成相似。
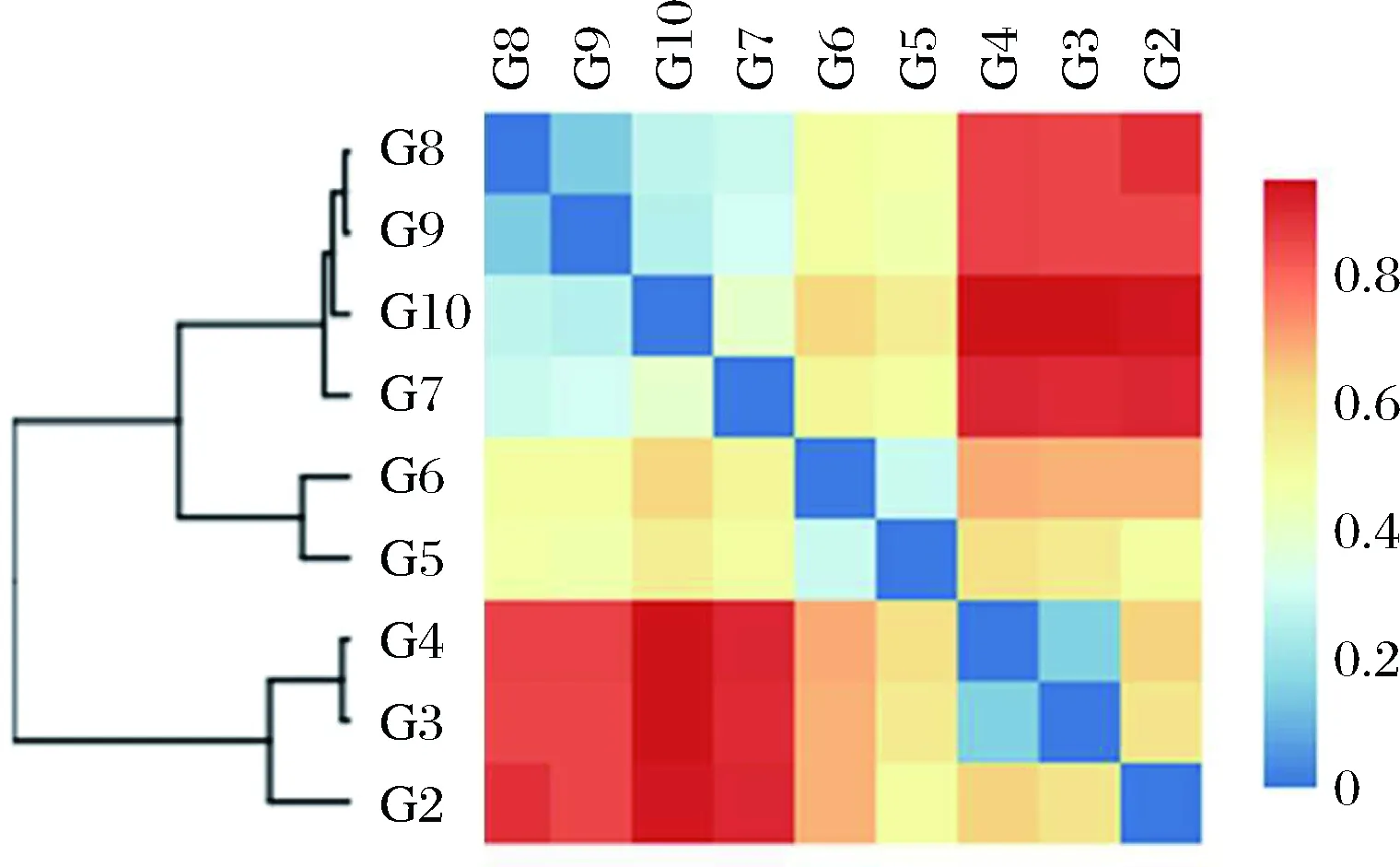
图4 样品Beta多样性热图Fig.4 Beta diversity heatmap of samples
3 讨论
通过传统分离培养方法,从勐海普洱茶发酵不同节点的9个样品中共分离得到18个属,30个种,其中优势菌属有微杆菌属(Microbacteriumsp.)、葡萄球菌属(Staphylococcussp.)、小短杆菌属(Brachybacteriumsp.)、考克氏菌属(Kocuriasp.)、泛菌属(Pantoeasp.)和芽胞杆菌属(Bacillussp.)等。16S rDNA序列分析发现这些优势属中分布有一定数量的新种,这在以往的普洱茶研究中尚未见报道。深入研究和挖掘这些新种在微生物分类学和发酵普洱茶微生物资源的保护方面具有积极意义。
通过高通量测序方法,9个样品共解析到细菌61个属,212个OTU,包含了培养方法分离到的菌种对应的属级分类单元,可见与传统方法相比,高通量测序更全面地揭示了发酵普洱茶样品的细菌群落构成。高通量测序数据分析显示:晒青毛茶原料的菌落构成较为丰富,包括乳杆菌属(Lactobacillussp.)、葡萄球菌属(Staphylococcussp.)、考克氏菌属(Kocuriasp.)等;而发酵前期以革兰氏阴性的欧文氏菌属(Erwiniasp.)、泛菌属(Pantoeasp.)和假单胞菌属(Pseudomonassp.)为主;而发酵中后期芽胞杆菌属(Bacillussp.)、葡萄球菌属(Staphylococcussp.)、短杆菌属(Brevibacteriumsp.)、考克氏菌属(Kocuriasp.)及小短杆菌属(Brachybacteriumsp.)为优势菌群稳定存在,约占样品群落组成的75%。
本研究对勐海百中堂茶厂普洱微生物的研究与其他学者的研究结论具有一定的一致性同时又有独特性。欧文氏菌、假单胞菌、芽胞杆菌、微杆菌等均为常见的植物内生菌,随着渥堆发酵的进行,温湿度、含氧量、原料基质理化特征发生变化,优势菌群也随之改变。王辉[12]等人对云南南涧凤凰沱茶厂发酵普洱茶的研究中发现:除了葡萄球菌属(Staphylococcussp.)、芽胞杆菌属(Bacillussp.)、短杆菌属(Brevibacteriumsp.)等优势菌外,欧文氏菌属(Erwiniasp.)在整个发酵不同阶段不同茶样层间均表现优势。而本研究仅在发酵前期发现欧文氏菌属(Erwiniasp.)占绝对优势(38%~58%)。姚静[11]等人通过分离培养法,发现芽胞杆菌属(Bacillussp.)在普洱茶发酵中所占比例远大于其他细菌,处绝对优势地位。葛慈斌[27]等人的研究发现多种芽胞杆菌是普洱茶发酵后期的优势菌群。芽胞杆菌能够适应不同的环境条件,可兼性厌氧生长,在高温缺氧条件下以芽胞的形式存活,因此在整个普洱茶发酵加工过程中优势存在。研究表明芽胞杆菌又是产生多种生物酶类的重要来源,其分泌的多酚氧化酶能够将黄烷醇类及其糖苷类底物转化为如双黄烷醇、茶黄素、茶红素等,在黑茶风味和成色的形成中起着重要作用[28]。葡萄球菌是发酵中后期的优势菌种,具有有氧呼吸和无氧发酵的能力,能提高过氧化氢酶活力,促进耗氧,抑制酸败微生物。据报道,S.carnosus、S.sciuri、S.xylosus、S.kloosii等葡萄球菌具有产大量支链醛、甲基酮和脂肪酸乙酯等芳香成分、硝酸盐还原和蛋白酶降解等能力[29-35]。据国际乳品联合会发布的欧美各国传统发酵食品中的微生物名单(IDF 455-2012),其中有多种葡萄球菌(S.carnosus、S.cohnii、S.condimenti、S.equorum、S.fleurettii、S.piscifermentans、S.saprophyticus、S.sciuri、S.succinus、S.vitulinus、S.warneri和S.xylosus)作为肉类、乳制品和豆类的发酵菌种,并有悠久的安全食用历史。本研究在发酵普洱茶中也分离到其中的S.sciuri和S.xylosus这2种葡萄球菌,其中S.xylosus也已于2016年被列入我国《可用于食品的菌种名单》。研究发现这些发酵食品中的葡萄球菌具有产芳香风味、硝酸盐还原和蛋白酶降解等能力[29-32]。本研究分离到的S.kloosii最早由SCHLEIFER等人于1984年发现并命名[36],在许多发酵食品,如豆豉[37]、豆酱[38]、Inyu(黑豆酱)[29]、Miso[39]发酵过程中均有分离到该菌种。陈廷涛发现在传统发酵豆豉中也有S.gallinarum[37]。本研究分离到的其他优势菌属在一些传统发酵食品中也有报道,如Kocuriakoreensis在韩国传统发酵海产品(comb pen shell)中被发现[40],Microbacteriumgubbeenense存在于斑点成熟奶酪的表层[41]。此外,Brachybacteriumzhongshanense报道具有纤维素分解能力[42]。其他优势菌种短杆菌属、微杆菌属、考克氏菌属菌种多以新种的形式存在,其在发酵过程中的作用有待进一步研究。鉴于安全性考虑,发酵普洱茶中的这些优势菌种的功能、酶学特性及安全性评价是下一步研究的重点。
猜你喜欢
中国人兽共患病学报(2024年2期)2024-03-15 02:41:52
河南医学研究(2022年19期)2022-10-19 00:44:18
中国生物防治学报(2022年3期)2022-07-09 10:00:22
微生物学杂志(2021年2期)2021-07-01 11:01:06
微生物学杂志(2020年2期)2020-12-31 07:17:13
生态学报(2019年11期)2019-07-08 06:18:58
动物营养学报(2015年10期)2015-12-01 02:26:34
食品工业科技(2014年23期)2014-03-11 18:19:08
华东理工大学学报(自然科学版)(2014年5期)2014-02-27 13:49:27
华东理工大学学报(自然科学版)(2014年5期)2014-02-27 13:49:27
